07-30-04 03:13
No 522581
We find on Rhodium site (../rhodium /halosaf
Aliquat 336 is trioctylmethylammonium chloride [(CH3)N(C8H17)3],Cl.
And in this document, we can also find that "(...) PTCs such as tetrabutylammonium bromide and triethylbenzyl-ammonium chloride serve as very poor PTC catalysts due to their appreciable solubility in water."
I found that didecyldimethylammonium chloride,
[CH3(CH2)9]2N(CH3)2,Cl is used as an anti-algae for swimming pools.
So I just wonder, (because I haven't find any data on this compound) : with its long alkyl chains, that OTC PTC will be relatively not water soluble, and its structure is not far from Aliquat 336, so maybe it can works as an OTC PTC for chlorosafrole preparation ? (Which can eventually be purified and swaped in acetone to iodosafrole...). Does someone ever tried that PTC, or any information about it, opinions ?
(Chief Bee)
07-30-04 04:36
No 522599
didecyldimethylammonium chloride [...] will be relatively not water soluble, and its structure is not far from Aliquat 336, so maybe it can works as an OTC PTC for chlorosafrole preparation ?
Yes, certainly! That PTC is likely to be relatively base-sensitive, but that is not an issue in this very acidic reaction.
The Hive - Clandestine Chemists Without Borders
(Stranger)
07-30-04 04:44
No 522601
But Rhodium, what will be wrong comparatively to Aliquat 336, with the acidity of the solution ?
(Chief Bee)
07-30-04 05:21
No 522613
My comment was superfluous and had nothing to do with your reaction.
The PTC you found is just fine for the PTC/HCl(aq) hydrochlorination of Safrole.
The Hive - Clandestine Chemists Without Borders
07-30-04 12:47
(Rated as: UTF PM Function)
(Hive Bee)
07-30-04 14:40
No 522692
Hmmmm, not bad....
So.......
As i see it the halosafrole route to MDMA and others was flawed by the use of iodosafrole, the yields for the halosafrole go up when you move from bromo to iodo but the yields go from ok to bupkis with the amine rxn....
Seeing as chlorosafrole forms lower yields (but now higher due to PTC) with the chlorosafrole formation would that mean the problem associated with the low yields of iodosafrole --> MDMA would be overcome by this?
The problem as i was informed was iodosafrole reactes with amines to form the salt of that amine and safrole... only forming the desired product in very low, if any yields...
Would chlorosafrole not do such a thing? or would the yield be greater enough to warrant it as a viable pathway?
Just thinking.....
-AC
Its just my opinion, but no-one listens to me anyway, and rightly so...
(Stranger)
07-30-04 15:11
No 522700
ApprenticeCook : the problem is now doing the amination, right ?
By two things must be resolved :
- The polyalkylation of the amine, because more alkyl groups it brings, more nucleophile it becomes.
- The use of iodosafrole, if you wanna have decent yield in your amination.
Here are two ideas that can, maybe, helps us :
For the fisrt problem, the use of a large excess of the ammine (ammoniac, methylamine... depending on what you want to do), can minimize the polyalkylation. (References in all good books).
For the second one, maybe, instead of doing a swap in acetone (chlorosafrole --> iodosafrole), we can just use a catalytic amount of an iodide salt, like Rhodium suggest in post 389608, to catalyse the substitution of Cl.
Opinions, or is something wrong with that ?
(Hive Bee)
07-31-04 03:41
No 522813
The problem with amine subst of iodosafrole is you only get mdma (or mda, mdea etc) in small yields...
You major yields are using for instance, iodosafrole and MeAm to make MDMA, MeAm iodide and safrole.
So by the addition of the amine your reversing your halogenation reaction...
Polyalkyl of the amine, yes far excess of amine is required to stop this. This is yet another hindering factor of this method.
Warm Acetone + NaI will subst chloro for iodo in excellent yields. But you have teh problem with amine as i said top of this post.
My question was in my first reply is that the point of chlorosafrole as an amination reactant would be interesting.... iodide is so reactive thats why it creates the amine salt better instead of HI and the desired alkylamine.
So if the yields are normally high for iododsafrole formation but low for the amination of iodosafrole, and the yields for the chlorosafrole formation are normally low (due to less reactivity of chloride, which your solving with the use of the PTC) wouldnt that low reactivity mean the yields of the amination of chlorosafrole be a bit better???
Also, whats the mechanism for the amination reaction of halosafrole? SN1 or SN2???
-AC
Its just my opinion, but no-one listens to me anyway, and rightly so...
(Hive Addict)
07-31-04 04:22
No 522828
The mechanism is SN2. Using the iodide has the downside of iodides poisoning PTC catalysts.
fear fear hate hate
(Stranger)
07-31-04 12:43
No 522915
Ok, I didn't know that there is the problem of the reformation of safrole and the formation of the iodide salt of the amine. Have you some references, or it's experimental result ?
So yes maybe it's more interesting to start from chlorosafrole for the amination, with the use of a PTC to increase a bit the yield. But the PTC that I speak about in my first post, didecyldimethylammonium chloride, works for the safrole / HCl(aq) reaction. I don't know if it will work for the amination.
But drone 342 posted a method for PTC amination (in TFSE, you will find), with benzyltriethylammonium chloride. So the idea might be to use two PTC, an Aliquat 336 analogue for the creation of halidesafrole, and benzyltriethylammonium chloride for the amination.
Concerning the fact that iodides poison the PTC, the first idea was just to use iodide as a catalyst for the amination, without PTC, but apparently iodosafrole won't react as I thought.
And what about putting, this time, a catalytic amount of a bromide salt instead of idodide ? Never heard about, I don't know if it may work.
And some interesting infos on PTC, look in "Issue" :
http://www.sacheminc.com/catalysts/ptc/p
(Chief Bee)
07-31-04 14:32
No 522923
Some comments about the extensive formation of emimination byproducts when aminating halosafroles with methylamine is mentioned in the following doc: ../rhodium /mdma.to
The tosylates used in the above doc reacts like "perfect halides", i.e. the substitution/elimination ratio is way more favorable. Also, as trans-isosafrole is thermodynamically more favored than either safrole and cis-isosafrole, the alkene which forms when a 2-halosafrole is eliminated is almost exclusively trans-isosafrole.
The Hive - Clandestine Chemists Without Borders
(Stranger)
07-31-04 15:40
No 522926
Relative low temp (ambiant or even fridge) can be used to minimize elimination.
Can ethanol/water mix with disolved amine in it be used for amination without PTC ? Since halosafrole are soluble in ethanol.
(Hive Bee)
08-01-04 11:46
No 523065
utfse for posts about the subject for refs im just speaking from trying it once and getting next to squat conversion....
utfse for iodosafrole and halosafrole, esp for posts by psychokitty and other long standing experimenters....
-AC
Its just my opinion, but no-one listens to me anyway, and rightly so...
(Stranger)
08-01-04 20:57
No 523104
I�ve searched with TFSE posts concerning halosafrole and particularly iodosafrole substitution. But I�ve not found comprehensive data concerning the competition between substitution and elimination.
I�ve searched in the Vollhardt �Organic Chemitry. Structure and Fonction� for precise details. So, chlorosafrole must follow Sn1, Sn2, E1 and E2 mechanism.
But �Bases that are weaker than hydroxide, but which are powerful nucleophiles (I- ; Br- ; N3- ; ...) give good yields in Sn2 with primary and secondary halides...� and �Weak nucleophiles, like water or alcool, react with a decent speed only with secondary and tertiary halide, with an Sn1 mechanism, with nearly no elimination.�
Moreover, elimination is minimized with low temp.
Ammonia is not a bad nucleophile (relative constant of 103 in methanol, water has 1, idide has 105) and it�s a weak base.
All of this is in favour of a dominant substitution reaction when reacting all types of halosafrole with ammonia, at low temp.
(Stranger)
08-02-04 20:53
No 523257
I seem to remember a thread from a few years ago when the halosafrole amination rxn (w/out a pressure-bomb) was being discussed extensively where someone gave a good reason why DMSO seemed to be by far the best solvent for this rxn... i cant remember exactly what the solvent property was at the moment, but DMSO seemed to be at the preferable end of the scale by several orders of magnitude, and it went sthng like DMSO >> DMF(i think?!) >> MeOH... hope that helps
-psyx
(Chief Bee)
08-03-04 03:28
No 523304
(Rated as: good read)
SN2 vs. E2
SN2 and E2 reactions share a number of similarities. Both require good leaving groups, and both mechanisms are concerted. SN2 reactions require a good nucleophile and E2 reactions require a strong base. However, a good nucleophile is often a strong base. Since the two reactions share many of the same conditions, they often compete with each other. The the outcome of the competition is determined by three factors: the presence of antiperiplanar β-hydrogens, the degree of α and β branching, and the nucleophilicity vs. basicity of the reactant species.
In order for an E2 elimination to occur, there must be antiperiplanar β-hydrogens to eliminate. If there are none, the SN2 reaction will dominate. On the same token, the SN2 nucleophile needs an free path to the σ* C-LG antibond. α and β branching block this path and reduce the proportion of SN2 relative to E2. E2 occurs even with extensive branching because it relies on the β-hydrogens, which are much more accessible than the σ* C-LG antibond.
The identity of the nucleophile or base also determines which mechanism is favored. E2 reactions require strong bases. SN2 reactions require good nucleophiles. Therefore a good nucleophile that is a weak base will favor SN2 while a weak nucleophile that is a strong base will favor E2. Bulky nucleophiles have a hard time getting to the α-carbon, and thus increase the proportion of E2 to SN2. Polar, aprotic solvents increase nucleophilicity, and thus increase the rate of SN2.
SN2
1. Requires an unhindered path to the back of the a carbon
2. α and β branching block the path and hinder SN2
3. Requires a good nucleophile
4. Polar, aprotic solvents increase nucleophilicity
5. Bulky groups on the nucleophile decrease nucleophilicity
E2
1. Requires an antiperiplanar β-hydrogen
2. Enhanced by α and β-branching
3. Requires a strong base
Source: Lu, Michael; SparkNote on Organic Chemistry: Sn2E2 Reactions (http://www.sparknotes.com/chemistry/org
Solvent Effects in Nucleophilic Substitution
In general terms, the choice of solvent can have a significant effect on the performance of a reaction.
Factors when chosing a solvent are:
- solubility: need to get reagents in the same phase, the molecules need to collide to react!
- usually, the solvent needs to unreactive towards the reagents (except in reactions where the solvent is the Nu: "solvolysis")
- how will the solvent affect the rate of reaction ?
For an SN1 reaction, the polarity and ability of the solvent to stabilise the intermediate carbocation is of paramount importance, as shown by the relative rate data for the solvolysis of tBuCl.
| Solvent | Dipole moment [�] |
Dielectric constant [ε] |
Relative Rate |
| CH3CO2H | 1.68 | 6 | 1 |
| CH3OH | 2.87 | 33 | 4 |
| H2O | 1.84 | 78 | 150,000 |
Dipole moment, � in debye
For an SN2 reaction, the effect of solvent polarity is usually much less, but the ability (or really lack there of) of the solvent to solvate the nucleophile is the important criteria, as shown by the relative rate data for the SN2 reaction of nBuBr with N3-.
| Solvent | Dipole moment [�] |
Dielectric constant [ε] |
Relative Rate |
Type |
| CH3OH | 2.87 | 33 | 1 | protic |
| H2O | 1.84 | 78 | 7 | protic |
| DMSO | 3.96 | 49 | 1,300 | aprotic |
| DMF | 3.82 | 37 | 2,800 | aprotic |
| CH3CN | 3.92 | 38 | 5,000 | aprotic |
Polar Protic Solvents (polar and ability to be H-bond donor)
- have dipoles due to polar bonds
- can H atoms that can be donated into a H-bond
- examples are the more common solvents like H2O and ROH
- remember basicity is also usually measured in water
- anions will be solvated due to H-bonding, inhibiting their ability to function as Nu
Polar Aprotic Solvents (polar but no ability to be H-bond donor)
- have dipoles due to polar bonds
- don't have H atoms that can be donated into a H-bond
- examples are acetone, acetonitrile, DMSO, DMF
- anions are not solvated and are "naked" and reaction is not inhibited
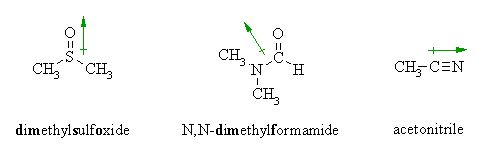
Overall
- All nucleophiles will be more reactive in aprotic than proticsolvents
- Those species that were most strongly solvated in polar protic solvents will "gain" the most reactivity in polar aprotic (e.g. F-).
- Polar aprotic solvents are typically only used when a polar protic solvent gives poor results due to having a weak Nu, (esp. F-, -CN, RCO2-)
Source: Francis A. Carey, Organic Chemistry, Ch. 8 - Nucleophilic Substitution (http://www.chem.ucalgary.ca/courses/351
The Hive - Clandestine Chemists Without Borders
(Stranger)
08-03-04 14:21
No 523370
The informations given by Rhodium summed up the theory of competition between E2 and Sn2. http://www.chem.ucalgary.ca/courses/351/
All that confirm the first idea :
- With a secondary halide, with one methyl group (accessibility is not really hindered),
- the use of (not a bad) nucleophile like NH3, which is a weak base (pKa = 9,2 and for information methylamine has a pKa of 10,6),
- the fact that NH3 isn�t an obstructed nucleophile,
- with a very good leaving group like I-
- at low temp,
the substitution will (clearly I think) predominate in a solvent like MeOH.
MeOH is a good solvent to my mind, because it can dissolve a large quantity of ammonia at low temp
(for ammonia solubility cf. http://www.larocheind.com/pdf/library/AT
Now we can ask ourselves what the influence of the use of a polar aprotic solvent could be. Will it increase significantly the nucleophility of NH3 ? And with that the yield of the reaction ?
(Stoni's sexual toy)
08-03-04 15:09
No 523374
> Now we can ask ourselves what the influence of the use of a polar aprotic
> solvent could be. Will it increase significantly the nucleophility of NH3 ?
Since NH3 isn't an anion the effect shouldn't be that pronounced as in the cases where the Nu is an anion. NH3 isn't that great as a nucleophile and it's also a weak base (both are closely related).
MeOH as the solvent isn't a good choice. The reaction will simply take too long. The same reaction in a dipolar-aprotic solvent will be many thousand times faster.
> And with that the yield of the reaction ?
What wasn't taken into account in the theoretical discussion presented in this thread is the proximity of the benzylic position and the relative ease it will release its hydrogen. This is the reason why allylbenzenes can be rearranged to propenylbenzenes while the reverse is impossible (or at least very difficult). This is also the reason why the elimination product will always be the propenylbenzene and not the allylbenzene. I have no literature to back this up but I think no matter what you do the elimination will always happen and fuck up your yield, at least when using the more common reagents.
I can't help it, I think people are wasting their times with this reaction and all its variations. It has been discussed countless times since the hive existed and no real progress has been made and most likely won't be made. Unless you consider other nucleophiles like azide this halosafrole + R-NH2 reaction sucks bigtime.
BUSH/CHENEY 2004! After all, it ain't my country!
www.american-buddha.com/addict.war.1.htm
(Hive Bee)
08-03-04 15:29
No 523377
Unless you consider other nucleophiles like azide this halosafrole + R-NH2 reaction sucks bigtime.
Unfortunatly to true....
The yields just are not high enough in any form to justify its use over peracid or wacker, and its actually a big pain in the ass to do the halosafrole route, when compared to the other availible methods.
-AC
Its just my opinion, but no-one listens to me anyway, and rightly so...