07-01-04 02:18
No 516633
Trimethylorthoformate from chloroform with NaOH/MeOH ? It just looks like. No NaOMe needed.
Am I the only one that is unable to see more than the first page of this patent ?
(www.espacenet.com doesn't work for me, I get all my patents from www.depatisnet.de)
Patent JP000058225036AA
PURPOSE: The reaction mixture resulting from a reaction between methanol or ethanol, a caustic alkali and chloroform is combined with a specific solvent to effect almost quantitative extraction of the titled compound into the solvent.
CONSTITUTION: Methanol, or ethanol, a caustic alkali, preferably an alcoholic solution of sodium or potassium hydroxide, and chloroform are made to react and the resultant reaction mixture is combined with a solvent which is selected from aliphatic or aromatic hydrocarbons, ethers or their halogenated products, and is low in compatibility with water and capable of separating the product by distillation, in an amount of 1.0W1.5 times the volume of the product. Then, they are vigorously stirred at 20W35°C and separated by standing to effect extraction of the objective orthoformate into the organic solvent layer, while the alkali chloride goes in the aqueous layer. The above solvent can be added at the reaction stage except carbon tetrachloride.
--psyloxy--
(Hive Addict)
07-02-04 00:46
No 516801
EDIT: a complete misunderstanding on my part.
Liebigs Ann.Chem., 9.1990;847-852
Ethyl (S)-2-(4-Tolylsulfonyloxy)propanoate (22): 4-Toluenesulfonyl chloride (6.0 g, 31.5 mmol) was added in portions to a solution of ethyl (S)-2-hydroxypropanoate (21) [2.5 g, 21.1 mmol; commercially available,[a]14 = -10° (neat); [a]20D = -24 (c = 8.59, CHCl32)] with vigorous stirring at - 20°C. Then the temperature was allowed to reach 0°C and stirring continued for 6 h, then for about 12 h at room temp. The mixture was poured into a mixture of ice/hydrochloric acid (1:1, 50 ml) and the organic layer was extracted with ethyl acetate. After drying the solution with sodium sulfate and evaporation of the solvent, 5.34g (93%) of 22 was obtained.

--psyloxy--
(A Truly Remarkable HyperLab Bee)
07-02-04 13:01
No 516946
Ethyl (S)-2-(4-Tolylsulfonyloxy)propanoate (22): 4-Toluenesulfonyl chloride (6.0 g, 31.5 mmol) was added in portions to a solution of ethyl (S)-2-hydroxypropanoate (21) [2.5 g, 21.1 mmol; commercially available,[a]14 = -10° (neat); [a]20D = -24 (c = 8.59, CHCl32)] with vigorous stirring at - 20°C.
Well, TsCl was added to a solution of ethyl lactate. A solution in what? Most probably, the authors meant a solution in pyridine, since they treat the reaction mixture with ice - HCl, a step which would be illogical if the reaction is conducted in the absence of a base. With pyridine as the solvent/base it becomes a standard tosylation of an alcohol.
I'm pretty sure that without a base the reaction of TsCl with a secondary alcohol is very slow, and only traces of the product will be formed after several hours at room temperature.
In the post Post 417550 (Antoncho: "Methyl tosylate: _finally_ , OTC!!!", Novel Discourse) methyl tosylate is made from TsCl - MeOH - conc. NaOH which form a biphasic system. The reaction takes place in the organic phase which contains almost no water, so the hydrolysis of TsCl is suppressed.
I don't know if there exists a PTC tosylation method compatible with esters.
(Hive Addict)
07-03-04 20:08
No 517202
(Rated as: excellent)
 Bull.Chem.Soc.Jpn., 68,1.1995;297-300 K2CO3 / Me3N.HCl
Bull.Chem.Soc.Jpn., 68,1.1995;297-300 K2CO3 / Me3N.HClpreparation of 2-Propynyl p-Toluenesulfonate 2a
To a stirred suspension of propargyl alcohol (5.6 g, 0.1 mol) and K2CO3 (15.2 g, 0.12 mol) in MIBK (50 mL) was added Me3N.HCl (0.96 g, 0.01 mol) at 0-5°C. After 5 min, p-toluenesulfonyl chloride (20.97 g, 0.12 mol) in MIBK (50 mL) was added at 0-5°C for an hout. After the mixture was stirred for an hour, water (100 g) was added at 20°C. The separated water phase was back extracted with MIBK (20 mL) and the combined organic extracts were first washed with water, and then, saturated aquaeous NaCl, dried (Na2SO4), concentrated, and purified by column chromatography (hexane/EtOAc=10:1) giving 19.13 g (91%) of the sulfonate 2a.
MIBK = methyl isobutyl ketone
Here's some other alternatives to Py / Et3N :
Bull.Chem.Soc.Jpn., 63,4.1990;1260-1262 NaOH / THFsynthesis 1999,9;1633-1636 TMEDA / Me2N-(CH2)n-NMe2 n=3, n=4, n=6 ;; shitty scan but you get the idea.Synthesis - 1997;1433-1438 DABCOTetrahedron Lett., 42.2001;8781-8783 Ag2O / KI--psyloxy--
(Chief Bee)
07-05-04 18:18
No 517576
(Rated as: excellent)
Preparation of Tosylates of Phenols and Acidic Alcohols
Stanley E. Wentworth and Patrick L. Sciaraffa, Organic Preparations and Procedures 1(4), 225-228 (1969)
(Article found by psyloxy & retrieved by azole)
Although there are many reports of the preparation of the title compounds1, each one differs somewhat from the next and the yields are variable.
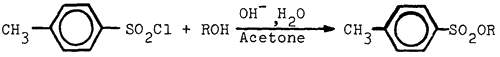
We wish to report a simple preparation of tosylates of phenols and of acidic alcohols. The method involves stirring an acetone solution of tosyl chloride and the alcohol (or phenol) with an excess of aqueous base, removal of the solvent, and isolation of the product. Reaction time is conveniently overnight, but could be reduced to as little as 4 h in the case of phenol without effect on the yield. The yields in many cases were better than those reported and were generally above 90%, except when a non-acidic alcohol such as n-butanol was used2. The low yield in this case may be due to further reaction of the tosylate with the excess base, or, as suggested by a referee, competitive hydrolysis of the tosyl chloride. Other compounds prepared by this method are reported in the Table.
Table
| Compound | Yield | mp (Found) | mp (lit.) |
| n-BuOTs | 50%a | - | - |
| C6H5OTs | 98% | 94-96 | 94-953 |
| p-ClC6H4OTs | 91% | 70-72 | 79.6-80.64 |
| p-BrC6H4OTs | 95% | 78-80 | 93-955 |
| p-IC6H4OTs | 95% | 97-99 | 996 |
| p-NO2C6H4OTs | 89% | 96-98 | 97-97.57 |
| CF3CH2OTs | 95% | 40-42 | 418 |
| C6H5CH(CF3)OTs | 94% | 113-116 | 113-1169 |
| C6H5CH2CH(CF3)OTs | 91% | 89-92 | b,d |
| C7F15CH2OTs | 75% | 53-56 | c,d |
| TsOCH2(CF2)3CH2OTs | 99% | 96-98 | 92-9410 |
a. Crude yield, not isolated.
b. New compound: Anal. Calcd. for C16H15F3O3S: S, 9.31. Found S, 9.19.
c. New compound: Anal. Calcd. for Ci5H9F1503S: C, 32.5; H, 1.63. Found: C, 32.4; H, 1.58.
d. The IR spectra of the new tosylates closely resembled those of the known tosylates (see fig.)
Experimental
Phenyl tosylate
To a stirred solution of 5.0g of tosyl chloride and 2.5g of phenol in 20 ml of acetone was added dropwise 1.28 g. of sodium hydroxide in 8 ml. of water. After having been stirred overnight, the solution was evaporated in vacuo. The resulting semi-solid was partitioned between ether and water. The layers were separated and the aqueous phase washed with a further portion of ether. The combined ethereal extracts were evaporated and the residue recrystallized from a mixture of hexane and acetone to give 6.45g (98%) of phenyl tosylate, mp 94-96°C, lit.3 mp 94-95°C.
References
1. R. B. Wagner and H. D. Zook, "Synthetic Organic Chemistry", John Wiley and Sons, Inc., New York, N. Y., 1953, p. 823.
2. A. T. Roos, H. Gilman, and N. J. Beaber, "Organic Syntheses", Coll. Vol. I, John Wiley and Sons, Inc., New York, N. Y., 1941, p. 145.
3. R. Otto, Ber. 19, 1832 (1886)
4. M. Neeman, A. Modiano, and Y. Shor, J. Org. Chem. 21, 671 (1956)
5. S. E. Hazlet, J. Am. Chem. Soc. 59, 287 (1937)
6. D. Matheson and H. McCombie, J. Chem. Soc. 1103 (1931)
7. E. Bamberger and A. Rising, Ber. 34, 228 (1901)
8. G. Van Dyke Tiers, H. A. Brown, and T. S. Reid, J. Am. Chem. Soc. 75, 5978 (1953)
9. R. A. Shepard and S. E. Wentworth, J. Org. Chem. 32, 3197 (1967)
10. B. S. Marks and G. C. Schweiker, J· Am. Chem. Soc. 80, 5789 (1958)
The Hive - Clandestine Chemists Without Borders
(Stranger)
08-17-04 15:21
No 525886
It would make sense that ROH + CHCl3 + [OH-] would produce orthoformates, to me at least--chloroform + hydroxide produces dichlorocarbene, which could insert into the alcoholic -OH group and produce CHCl2OR. However, when this happens, another HCl can become eliminated, forming another carbene, and inserting again to produce CHCl(OR)2.
The process would repeat a third and last time to yield CH(OR)3, trialkyl orthoformate. As long as the formed carbenes reacted with alcohol better than with the water formed by neutralization of base by the eliminated HCl, it would work.
I guess this is a similar process to how diazomethane inserts itself into hydroxyls. The cool thing is that it doesn't need such a strong base, and there is no competition between substitution and elimination reactions, since both have the same outcome!
It's good to bee back! Don't trust your computer!!