Summary
The desmethyl compound, 4-hydroxy-N-methyltryptamine (4), was synthesized via a four-step reaction sequence starting from 4-benzyloxyindole (1). Psilocin (4-hydroxy-N,N-dimethyltryptamine), an indole hallucinogen, was labeled by N-monomethylation of the side chain using the classical methylating agent [11C]CH3I in 20±5% (decay corrected from [11C]CH3I) radiochemical yield. The radiosynthesis, semi-preparative HPLC and formulation were completed in an average time of 45 minutes.
Introduction
Fig. 1.
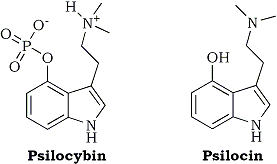
A considerable number of compounds possessing the indole moiety have gained much interest in recent years because of their hallucinogenic actions in man. Psilocin (4-hydroxy-N,N-dimethyltryptamine) and its O-phosphorylated derivative psilocybin (Fig. 1) are hallucinogenic and stand unique because of the unusual substitution at the 4-position of the indole ring. Both compounds were first isolated by Hofmann1 from the mushroom Psilocybe mexicana, a sacred mushroom used in pre-Columbian cultures by the natives in South America for religious purposes. The minimal side-effects and the short duration of action of psilocin and psilocybin have made possible their widespread use as psychotic drugs and as models in research to investigate psychiatric disorders such as acute states of schizophrenia. The fact that psilocybin produces a psychosis-like syndrome in man and the finding that frontocortical glucose metabolism in normal subjects similar to what is seen in acutely ill schizophrenics, suggests that psilocybin is a useful molecular probe to study the pathophysiology of psychotic disorder2. Moreover, there is a growing body of evidence from functional animal studies suggesting that LSD (lysergic acid diethylamide) and psilocybin exert their psychological effects through excessive 5-HT2 receptor activation3,4 while 5-HT2 receptor abnormalities have been reported in postmortem schizophrenic brain5-8. Numerous studies in experimental animals have shown that psilocybin is rapidly and extensively O-dephosphorylated by alkaline phosphatase enzyme to psilocin. These results suggest that psilocybin is a prodrug and that the pharmacologically active compound acting centrally is the 4-OH metabolite, psilocin9-12. This is reasonable in view of the fact that psilocybin, due to its low lipid solubility, probably does not pass the blood-brain barrier11. Furthermore, in experimental animals the same behavioral changes were produced using either psilocin or psilocybin.
A number of studies with LSD and a series of indole hallucinogens including psilocin suggest that these compounds may act as 5-HT2 agonists at the postsynaptic 5-HT2 receptors13. In the rat cortex, psilocin displayed a Ki value of 12±3 nM for the 5-HT2A receptor subtype14. Psilocin, in contrast to LSD, displays only negligible affinity (IC50>10000 nM) for the dopamine receptors15. The possibility to use [11C]psilocin for PET experiments might throw more light on the role of serotonin receptors in psychotic symptom formation. As part of our program on hallucinogenic compounds, we wish to report here on the synthesis and carbon-11 radiolabelling of psilocin.
Results and Discussion
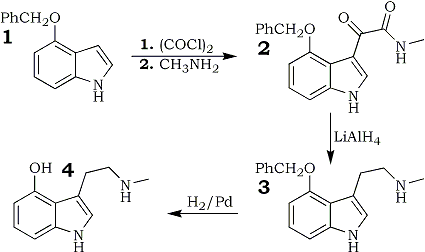
Fig. 2.
Synthesis of 4-hydroxy-N-methyltryptamine (4)
The reaction scheme for the synthesis of 4-hydroxy-N-methyltryptamine (4) is shown in Fig 2. 4 was synthesized using a four-step reaction sequence. The synthetic approach was an adaptation with slight variations of the procedures described in the literature1,16.
The starting material 1 which was protected at the 4-hydroxy position with the benzyl group allowed a selective reaction at the 3-position of the indole ring with oxalyl chloride. The reaction of 1. with oxalyl chloride and methylamine and the subsequent reduction of the intermediate glyoxylamide 1 with lithium aluminium hydride were accomplished in good yields. The benzyl group was easily removed by hydrogenolysis using Pd/C as catalyst at atmospheric pressure. However, due to the instability and exceedingly air-sensitive nature of the resultant 4-hydroxy-N-methyltryptamine rather low yields were obtained and the purification step following the hydrogenation remains to be a crucial part in the whole synthesis.
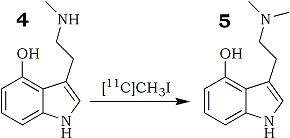
Fig. 3.
Radiosynthesis of [11C]psilocin (5)
The radiosynthesis of [11C]psilocin (Fig. 3) was accomplished by the N-methylation of the desmethyl precursor, 4-hydroxy-N-methyltryptamine, using the classical methylating agent [11C]methyl iodide. [11C]Methyl iodide was obtained from the gas phase reaction of [11C]methane with iodine. The yields of [11C]iodomethane achieved by this method are comparable to those reported recently17.
In order to obtain reasonable yields of [11C]psilocin it was necessary to prepare the free amine base under inert atmosphere. Optimal reaction conditions were accomplished using acetonitrile as solvent, a reaction temperature of 80°C and a reaction time of 10 min. The success of the radiolabeling depended much on the purity of the desmethyl compound and the mobile phase used for the purification. Very low yields (<10%) were obtained when the purity of the desmethyl precursor was below 70%. Solvent compositions which required evaporation prior to product formulation could not be used because [11C]psilocin obtained after purification was significantly decomposed on heating (Fig 4). Thus, the best eluent was a mixture of 0.06M H3PO4/ethanol (95:5) which was directly used for intravenous injection. The radiosynthesis, semi-preparative HPLC and formulation were completed in an average time of 45 minutes with radiochemical yield of 20±5% (decay corrected from [11C]CH3I). Specific activities obtained ranged from 900-2300 Ci/mmol at EOS. [11C]Psilocin, synthesized according to this procedure was characterized by 13C-NMR and MS using positive ion mode with electrospray as interface. The 13C signal at 43.9 ppm corresponded to the N-methyl group of authentic psilocin. Using mass spectroscopy molecular ion peaks at m/z 206 (M+1) and 205 (M+1) were observed for 13C-enriched psilocin and authentic psilocin, respectively. The radiochemical purity of [11C]psilocin was higher than 97% (Fig. 4). [11C]Psilocin was radiochemically stable at room temperature in phosphate buffer solution containing ascorbic acid over the period of use.
In conclusion, we have successfully synthesized and radiolabeled psilocin with carbon-11. The radiochemical yield and the specific activity are high enough to allow PET investigations of the role of serotonin receptors in psychotic symptom formation in humans. In vivo experiments are underway and will be reported elsewhere.
Experimental
Acetonitrile, oxalyl chloride, methanol, methylamine, lithium aluminium hydride (LiAlH4), 10% Pd/C, diethyl ether (distilled from LiAlH4 and dried over molecular sieves, 4Ĺ) and 4-benzyloxyindole were purchased from Fluka. Psilocin was obtained as a gift from Sandoz AG, Basle, Switzerland. Mass spectrometric analyses unless otherwise stated were performed using Fisions VG Autospec Q MS-system. IR spectra were measured on a Perkin Elmer 1600 series Spectrophotometer. NMR-spectra were obtained on Bruker Spectrospin AM 400 or Bruker AC-250 using tetramethylsilane as an internal standard. Melting points were determined on Büchi 530 apparatus (Büchi Switzerland) and are uncorrected.
Two systems were used for the isocratic HPLC separations:
System A (semi-preparative): Consisting of Waters 510 pump, a valco 6-port with a 10 mL loop, a Merck L-4000 UV detector (at 254 nm) a Geiger-Müller counter LND 714 with an Eberlein RM-14 instrument and a Knauer column Lichrosorb RP18 (250x10 mm, 10 µm) with 0.06M H3PO4/ethanol (95:5) at a flow rate of 2.5 mL/min.
System B (analytical): Consisting of Rheodyne injector with 100 gm loop, a Merck-Hitachi L 6200 pump, a NaI scintillation detector (Scintillation Meter type 540, Mini Instruments Ltd, Burnham on Crouch/UK), a Merck L-4000 UV detector (at 254 nm), a Merck-Hitachi D-2500 Chroma integrator, a Bondclone C18 column, 300x3.9 mm with 0.1% H3PO4:MeCN (3:2) at a flow rate of 0.8 mL/min.
(4-Benzyloxy-3-yl)-N-methylglyoxylamide (2)
As previously reported16 116.4 mmol of 4-benzyloxyindole (1) were dissolved in 250 mL of dried diethyl ether, cooled to 2°C and 232.8 mmol of oxalyl chloride in 200 mL of dried diethyl ether were added. The orange mixture was stirred for 1 hour, cooled (-10°C) and methylamine gas was passed into the reaction mixture until the orange colour disappeared. After stirring for an additional 1 hour at room temperature the precipitated materials were collected and washed with diethyl ether and water. The solid was dissolved in dichloromethane and water. The organic phase was separated, dried over Na2SO4, filtered and concentrated under reduced pressure to give 23.0g (64%) of 2.
Compound 2 was dissolved in a small amount of refluxing methanol/toluene (1:1). Precipitation with petroleum ether gave 57% of pure 2. with a melting point of 167°C (lit. 167-168°C): Spectral data identical to those prepared by the literature method16.
(4-Benzyloxy-3-yl)-N-methyltryptamine (3)
A solution of 40.2 mmol 2 in 200 mL abs. dioxane was added slowly under nitrogen to a boiling solution of 402 mmol LiAlH4 suspended in 200 mL abs. dioxane. The mixture was stirred for 18 hours at reflux and excess LiAlH4 was decomposed by the addition of methanol and saturated Na2SO4 solution at 0°C. The precipitated material was removed by filtration and the filtrate was concentrated under reduced pressure. The residue was dissolved in dichloromethane and made basic with 10% ammonia. The organic phase was separated, dried (Na2SO4) and evaporated. The crude product was purified by flash chromatography using a solvent system consisting of CH2Cl2/CH3OH/6M NH3 (20:80:10) to give 6 g (53%) of 3. IR and NMR data are identical to that reported in the literature16.
4-Hydroxy-N-methyltryptamine (4)
To a suspension of 1.5 g Pd/C (10% Pd) in 150 mL methanol (degassed) 10 mmol of 3 dissolved in 100 mL methanol were added dropwise. The mixture was stirred for 24 hours under H2 atmosphere at room temperature. The catalyst was removed by filtration under nitrogen and the filtrate was concentrated under vacuum to give 1.37 g (72%) of crude product which was 60% pure by GC-MS. 50% acetic acid (20 mL) was added to the crude product and the resultant acetate salt was washed with CH2Cl2. Purification by flash chromatography (silica gel, 60 mesh) using methanol: n-propanol:acetic acid (20:12:1) as eluent and lyophilization gave 0.4 g (21%) of 4 as the acetate salt which was 92% pure by GC-MS. The NMR and MS spectral characteristics of the acetate salt were in accordance with the structure of 4.
1H-NMR: (CD3OD, 400 MHz): δ 2.40 (s; 3H, N-CH3), 2.95 (t, J = 6.3 Hz, 2H, CH2-α/CH2-ß),3.05 (t, J = 6.4 Hz, 2H, CH2-α/CH2-ß), 4.90 (br. OH, NH, H2O), 6.33 (dd, J = 7.4/0.9 Hz, 1H, H-5), 6.80 (dd, J = 8.0/0.9 Hz, 1H, H-7), 6.86 (dd, J=7.4/8.0 Hz, 1H, H-6), 6.88 (s, 1H, H-2).
13C-NMR: (CD3OD, 400MHz): δ (ppm) = 27.10 (CH2), 35.44 (N-CH3), 53.60 (CH2-N), 103.94 (C-5/C-7), 105.00 (C-5\C-7), 113.50 (C-3/C-9), 118.52 (C-3/C-9), 122.21 (C-2/C-6), 123.35 (C-2/C-6), 140.73 (C-8), 153.27 (C-4).
MS: m/z = 190 (30%, M+), 147 (100), 146 (84), 118 (16), 117 (16), 44 (86).
Radiosynthesis of [11C]CH3I
[11C]CH3I was prepared from [11C]CO2 according to the standard procedure described in the literature17,18.
Radiolabelling of [11C]psilocin
The acetate salt of norpsilocin (2 mg, 0.008 mmol) was suspended in 400 µL of dry acetonitrile and 4 µL 5M NaOH were added. The mixture had a light blue colour and was stirred at room temperature under nitrogen for at least 5 minutes. The solution was then transferred under nitrogen atmosphere via syringe into a vial which had been oven-dried (150°C) previously. The reacti-vial was then positioned in a quartz lamp-heated vessel and [11C]CH3I was added to the solution via a slow stream of nitrogen. Following the complete addition of [11C]CH3I the flow of nitrogen was stopped and the reaction vessel was heated to 80°C for 10 min. Purification was achieved by a semi-preparative reversed-phase HPLC using a C-18 Lichrosorb column (250x10 mm, 10 µm) with 0.06M H3PO4/ethanol (95:5) as mobile phase at a flow rate of 2.5 mL/min. The desired product eluted between 16 and 19 minutes and was collected after filtering through a Millipore filter (0.22 µm) into a penicillin flask containing 100 µL (1 mg/mL) of L-ascorbic acid and 1.5 mL of 0.6 M phosphate buffer.