(Hive Bee)
05-16-03 20:18
No 433647
(Rated as: excellent)
This post is essentially an excerpt of the PhD dissertation Synthesis and Pharmacology of non-traditional cannabinoids. In its native form it is far from concise, so I've tried to cut it down, and although much of it is still in the authors own words, I have taken some liberties and changed the layout. Also, there are errors in the orginal document, and I have corrected a few, (I may have introduced typos and other errors too), so if anything looks suspect don't hesitate to post/PM
Synthesis and Pharmacology of non-traditional cannabinoids. A Dissertation by Jianzhong Lu (December 2000)
Presented to the Graduate school of Clemson University
The dissertation looks at four types of cannabinoids:
1 - cannabimimetic indoles
2 - a delta8-THC derivative with the side chain modified
3 - a hybrid cannabinoid which combines the structural features of both groups of active compounds
4 - a pyridone analogue of anandamide
1) Cannabimmetic Indoles:
Two groups of derivatives were prepared: 2-ethyl-3-(1-naphthoyl)indoles, which turned out to have little affinity for the cannabinoid brain receptor (CB1). Another group which was prepared was indole derivatives with a methoxy group substituted in the naphthoyl moiety at the 6- and 7- positions. Pharmacology data showed that the indole derivatives with a methoxy group at C-7 have better affinity than the C-6 isomers, and some of them have better affinity than the prototypical cannabinoid (delta9-THC).
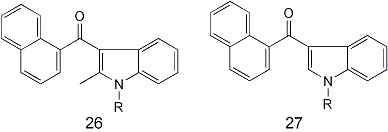
26 and 27 (27 = 1-alkyl-3-(1-napthoyl)indoles) 1-butyl, 1-pentyl and 1-hexyl derivatives all show typical cannabinoid pharmacology, with a full spectrum of dose related effects in the mouse.
Series 27 (unsubtituted at the indole 2-position) have generally have greater affinity for CB1 (brain cannabinoid receptor), while series 26 has greater affinity for CB2 (spleen cannabinoid receptor),33 1-alkyl-2-methyl-3-(7-methyl-1-naphthoyl
Affinity for the CB1 recpetor follows the pattern which was observed for indoles 26 and 27 (i.e. 1-methyl and 1-ethyl analogues are inactive: affinity for the CB1 receptor increases from R = n-propyl to R = n-pentyl then decreases when R = n-heptyl.
A 7-methyl substituent on the naphthalene group (33) has little effect upon the affinity for the CB1 receptor, with the exception of 33, R = n-heptyl) which has slight activity, whereas the analogue lacking the methyl group (26,R = n-heptyl) is completely inactive. The C-2 unsubstituted 4-methoxy-1-naphthoylindole analogues (34) have somewhat higher affinity for the CB1 receptor than the corresponding unsubstituted naphthoyl anaogues 27, but show only slight selectivity for the CB2 receptor31. The 1-n-pentyl analogue of 34 has the greatest affinity for the CB1 receptor of any of the cannabimimetic indoles reported to date by the Huffman group (Ki = 1.2+/-0.1 nM) and exhibits corresponding potency in vivo. The 2-methyl series (35) in general has slightly less affinity for the CB1 receptor than C-2 unsubstituted indoles 34.
On the basis of modeling studies (Huffman 1994) it was concluded that the aminoalkyl group characteristic of the Winthrop aminoalkylindoles is not necessary for canabinoid activity. This hypothesis led to the design and synthesis of a number of 1-alkyl-3-(1-naphthoyl)indoles (26, 27) some of which are very potent cannabinoids, both in vitro and in vivo. In particular, 1-butyl through 1-hexyl indole analogues, either unsubstituted at C-2 in series 27, or with a 2-methyl group in series 26, have affinities for the CB1 receptor in the ranges of 10 - 48 nM. The in vivo potencies of these cannabimimetic indoles are consistent with their receptor affinities30.
Subsequent experiments using mutant CB1 receptors suggest that traditional cannabinoids and cannabimimetic indoles bind to different , but partially overlapping sites on the receptor41,42.
These mutant receptor experiments, combinded with modeling studies have led to the hypothesis that the hydrogen bonding interaction of a lysine on the third transmembrane domain of the CB1 receptor is important in the binding of traditional cannabinoids, such as delta9-THC, and presumably other traditional cannabinoids43,44. However, the direct alignment of indoles and delta9-THC which led
to the development of a number of potent cannabimimetic indoles derived from 26 and 27 implies that there is a possibility that a single pharmacophore for both classes of cannabinoids could be developed29,30,45.
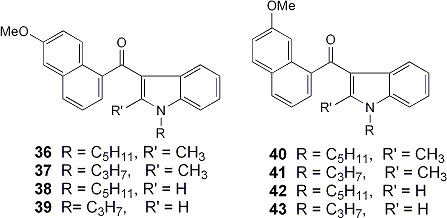
Indole compounds 28-32 with an ethyl group at C-2, were designed in a search for selective ligands for the CB2 receptor, while indoles 36-39 with a methoxy group at C-6 of the naphthol ring and 40-43 with a methoxy at C-7 were designed to explore the SAR for cannabimimetic indoles at both receptors.
Outline of synthesis of 2-Ethyl-3-(1-naphthol)indole derivatives
Based on Augustine et al.50 the reaction of 2-nitrobenzaldehyde with 1-nitropropane in the presence of anhydrous ammonium acetate gave nitrostyrene 51 This compound was reduced by catalytic hydrogenation at 50 psi in a mixture of ethyl acetate, acetic acid and absolute ethanol to give 2-ethylindole (52) as a gummy oil in 32% yield.
Indole 52 was treated with EtMgBr and the organometallic intermediate was treated with 1-naphthoyl chloride.
The crude product was heated at reflux with NaOH in MeOH which served to hydrolyze the N-acylated product to provide 3-naphthoylindole 53. N-alkylation29 using different alkyl bromides in the presence of KOH in DMSO at 80° C provided the final products (28-32) in yields from 79% to 95%
Pharmacological data
| Compound No. | R | R' | Yield (%) | CB1 affinity (Ki in nM +/-SEM |
| 28 | n-propyl | 79 | 2708+/-541 | |
| 29 | n-butyl | 88 | 203+/-13 | |
| 30 | n-pentyl | 89 | 52+/-5 | |
| 31 | n-hexyl | 89 | 272+/-84 | |
| 32 | n-heptyl | 95 | 1245+/-58 | |
| 36 | n-C5H11 | CH3 | 60 | 250+/-24 |
| 37 | n-C3H7 | CH3 | 81 | >10,000 |
| 38 | n-C5H11 | H | 55 | 44+/-10 |
| 39 | n-C3H7 | H | 56 | 2358+/-82 |
| 40 | n-C5H11 | CH3 | 43 | 45.0+/-0.8 |
| 41 | n-C3H7 | CH3 | 65 | 1568+/-201 |
| 42 | n-C5H11 | H | 66 | 6.6+/-0.7 |
| 43 | n-C3H7 | H | 64 | 204+/-26 |
Pharmacological evaluation of 28-32 showed that these compounds have weak affinities for the CB1 receptor (Ki = 51-2708 nM). On the other hand, the cannabimimetic indoles in series 26 and 27 have better affinities for the CB1 receptor (Ki = 9.5-164 nM for 26, Ki =9-1050 for 27. It seems substituents larger than methyl at C-2 of indoles will attenuate CB1 affinity.
The CB1 receptor affinities of the indole derivatives with a methoxy group at C-6 are summarized in Table 2.2 and those with a methoxy group at C-7 in Table 2.3. Compound 41 has the best affinity for the CB1 receptor (Ki = 6.6+/-0.7 nM). It is interesting to notice that all those indole compounds with the methoxy group at C-7 are more potent than the isomers with the methoxy group at C-6. Some of the indole derivatives in this series are more potent than the natural product delta9-THC.
Experimental:
1-(2-Nitrophenyl)-2-nitrobutene (51):
A mixture of 0.50g (3.3 mmol) of 2-nitrobenzaldehyde, 3mL of nitropropane and 0.13g (1.7 mmol) of anhydrous ammonium acetate was refluxed for 6 h. After cooling, the solution was diluted with 60mL of water and extracted with ethyl acetate. The organic solution was washed with brine three times and dried (MgSO4). The solvent was removed in vacuo and the residue was purified by column chromatography (petrolum ether/ethylacetate 8:1) to give 0.53g (72%) of product as a yellow oil.
2-Ethylindole (52):
To a solution of 0.52g (2.7 mmol) of 51 in 40mL of ethyl acetate, was added 1mL of absolute ethanol, and 1.2mL of acetic acid followed by 0.07g of 10% Pd/C. The mixture was shaken under an atmosphere of H2 at 50 psi and room temperature for 18 h. The catalyst was filtered off and the solvent was removed in vacuo.
The residue was purified by column chromatography (petrolum ether/ethylacetate 8:1) to give 0.11 g (32%) of product as a viscous oil.
2-Ethyl-3-(1-naphthoyl)indole (53):
To a stirred solution of 0.65mL (2.5 M, 1.65 mmol) of EtMgBr in ether diluted with 1.1mL of ether at 0° C under nitrogen, a solution of 0.20g (1.3 mmol) of 2-ethylindole (52) in 1.1mL of ether was added dropwise. The solution was kept stirring for 0.5 h, while the temperature was allowed to rise from 0° C to room temperature. A solution of 0.22mL (1.46 mmol) of 1-naphthoyl chloride in 1.0mL of ether was added dropwise, and the reaction mixture was stirred for 1.5 h. The reaction was quenched with saturated NH4Cl solution and stirred until the solid was broken up into a fine suspension. The solvents were removed by filtering all the mixed aqueous and organic solutions. The residue was washed with water, and ether, then suspended in 5mL of methanol to which was added 0.4g (10 mmol) NaOH and 1.0mL of water. The mixture was stirred overnight, filtered, and washed with successive portions of methanol, water and ether. The solid was fried in a vacuum oven at 100° C to give 0.31g (74%) of product as a viscous oil.
2-Ethyl-3-(1-naphthoyl)-1-propylindole (28):
To a solution of 0.40g (1.3 mmol) of 53 in 3.0mL of DMSO, 1.20g (30.8 mmol) of powdered KOH was added. The reaction was stirred for 1 h at room temperature, then 1.35mL (11.0 mmol)of bromopropane was added slowly. The solution was heated to 85° C and stirred overnight. After cooling it was diluted with water, extracted with ethyl acetate (3 x 35mL); the organic layer was washed with three portions of brine and dried (MgSO4).
The ethyl acetate was removed in vacuo and the reside was purified by column chromatography (petroleum ether/ethyl acetate 7:1) to give 0.36 g (79%) of a product as a yellow oil.
| 53 | Haloalkane | Product | Yield |
| 0.40g (1.3 mmol) | bromopropane - 1.35g (11.0 mmol) | 2-Ethyl-3-(1-naphthoyl)-1-propylindole 28 | 0.36g (79%) |
| 0.40g (1.3 mmol) | bromobutane - 1.20g (30.8 mmol) | 2-Ethyl-3-(1-naphthoyl)-1-butylindole 29 | 0.42g (88%) |
| 0.40g (1.3 mmol) | bromopentane - 1.20g (30.8 mmol) | 2-Ethyl-3-(1-naphthoyl)-1-pentylindole 30 | 0.44g (89%) |
| 0.40g (1.3 mmol) | bromohexane - 1.90mL (10.0 mmol) | 2-Ethyl-1-hexyl-3-(1-naphthoyl)indole 31 | 0.49g (89%) |
| 0.40g (1.3 mmol) | bromoheptane - 2.10mL (10.0 mmol) | 2-Ethyl-1-hepyl-3-(1-naphthoyl)indole 32 | 0.49g (95%) |
All products were yellow oils
2-Methyl-1-pentylindole (59):
To a solution of 1.02g (7.8 mmol) of 2-methylindole in 5.0mL of DMSO, 2.10g (37.5 mmol) of powdered KOH was added. The reaction was stirred for 1 h at room temperature, then 3.0mL (24.0 mmol) of bromopentane was added slowly. The solution was heated to 70° C and stirred overnight. After cooling it was diluted with water, extracted with ethyl acetate (3 x 35mL); the organic layer was washed with three portions of brine and dried (MgSO4). The ethyl acetate was removed in vacuo and the reside was purified by column chromatography (petroluem ether) to give 1.30g (85%) of product as a yellow oil.
| Starting Material | Haloalkane | Product | Yield |
| 2-methylindole - 1.02g (7.8 mmol) | Bromopentane - 3.0mL (24.0 mmol) | 2-Methyl-1-pentylindole 59 | 1.30g (85%) |
| 2-methylindole - 1.02g (7.8 mmol) | Iodopropane - 3.5mL (35.0 mmol) | 2-Methyl-1-propylindole 60 | 1.23g (93%) |
| Indole - 1.02g (8.1 mmol) | Bromopentane - 3.1mL (24.3 mmol) | 1-Pentylindole 61 | 1.15g (71%) |
| Indole - 1.02g (8.1 mmol) | Iodopropane - 3.5 mL (35 mmol) | 1-Propylindole 62 | 1.12g (82%) |
All products appeared as a yellow oil.
Got democracy?http://www.dhushara.com/book/multinet/de
(Hive Bee)
05-16-03 20:39
No 433650
[part two]
3,4-Dihydro-6-methoxy-1-tetralone triflate (63):
To a solution of 0.56mL (4.0 mmol) of N,N-diisopropylamine in 5mL of THF, 1.6mL (2.22 M, 3.6 mmol) of n-BuLi in hexane was added dropwise at 0° C. After 10 min the solution was cooled to -78°C and stirred for 15 min, and a solution of 0.5g (2.9 mmol) of 6-methoxy-1-tetralone in 7mL of THF was added to the above solution over a period of 15 min. The reaction was stirred for 30 min at -78°C, then allowed to warm to room temperature and stirred for 2 h. After cooling to -78° C, 1.12 (3.2 mmol) of Nphenyltrifluoromethanesulfonimide in 7mL of THF was added. The solution was warmed to 0° C, quenched with water and extracted with ether. The ethereal extracts were washed successively with and brine, and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed (petroleum/ether/ethyl acetate 12:1) to give 0.73g (85%) of product as a colorless oil.
Similarly, 3,4-Dihydro-7-methoxy-1-tetralone triflate65 may be prepared with the use of 0.5g (2.9 mmol) of 7-methoxy-1-tetralone, affording 0.53g of the triflate (61%)
3,4-Dihydro-6-methoxytetralone-1-carboxy
To a solution of 0.48g (1.6 mmol) of triflate 63 in 6mL of DMF, 13mg (0.15mmol) of triphenylphosphine was added, followed by 17mg pf palladium (II) acetate (0.075 mmol), and 0.6mL (4.31 mmol) of triethylamine. The mixture was flushed with carbon monoxide for 20 min, then 1.6mL of 96% formic acid was added and the reaction was stirred under a carbon monoxide atmosphere for 20 h. The mixture was diluted with water and extracted with ethyl acetate. The combined ethereal extracts were washed with brine, water and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed (petroleum ether/ethyl acetate 4:1) to give 0.25g (77%) of product as colorless needles: m.p. 132-134° C.
3,4-Dihydro-7-methoxtetralone-1-carboxyl
To a solution of 0.87g (2.8 mmol) 7-methoxytetralone triflate 65 in 6ml of DMF. 75mg (0.15 mmol) of triphenylphosphine was added, followed by 32mg of palladium (II) acetate (0.14 mmol), and 1.0mL (7.19 mmol) of triethylamine. The mixture was flushed with carbon monoxide for 20 min, then 1.6mL of 96% formic acid was added and the reaction was stirred under a carbon monoxide atmosphere for 20h. The mixture was diluted with water and extracted with ethyl acetate. The combined ethereal extracts were washed with brine, water and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed (petroleum ether/ethyl acetate 12:1) to give 0.46g (79%) of product as colourless needles. m.p 119-120° C
2-Bromo-6-methoxy-1-tetralone 67:
A solution of 2.0g (12.5 mmol) of bromine in 5.5mL of carbon tetrachloride and 0.5mL of dry ether was added dropwise (each drop was added only after the previous drop was completely decolorized) to a stirred solution of 2.2g (12.5 mmol) of 6-methoxy-1-tetralone in 50mL of dry ether, which contained 1mL of ethereal HCl. The solution was maintained at 0-5° C throughout the addition. After the addition of bromine was complete, the reaction mixture was stirred for 1 h and quenched with water. The mixture was extracted with ether, washed with water and dried (MgSO4). Removal of solvent afforded 3.1g (97%) of the expected bromo compound. The product was recrystallized from ether/petroleum ether (2:1) to give 2.5g (79%) as cubic crystals, m.p. 75-77° C.
| Tetralone | Product | Yield | M.p./appearance |
| 6-methoxy-1-tetralone 2.2g (12.5 mmol) | 2-Bromo-6-methoxy-1-tetralone 67 | 2.5g (79%) | 75-77° C cubic crystals |
| 7-methoxy-1-tetralone 4.0g | 2-bromo-7-methoxy-1-tetralone 75 | 5.0g (86%) | 75-77° C cubic crystals |
6-methoxy-1-naphthol 68:
To a solution of 2.0g (7.84 mmol) of 2-bromo-6-methoxy-1-tetralone in 40mL of DMF, 1.6g of lithium bromide (18 mmol) and 1.2g of lithium carbonate (17.4 mmol) were added. The mixture was refluxed for 6 h under N2, cooled, and diluted with water and ether. The ethereal solution was washed with three portions of brine, followed by 10% aqueous NaOH solution (3 x 100mL). The alkaline extracts were acidified and extracted with ether. Removal of the ether afforded 1.35g (99%) of crude naphthol, which was recrystallized from ethyl acetate/petroleum ether (2:1) to give 1.2g of pure 68, m.p. 85.5-86.5° C.
| Bromo-tetralone | Product | Yield (g) | M.p./appearance |
| 2-bromo-6-methoxy-1-tetralone, 2.0g (7.84 mmol) | 6-methoxy-1-naphthol 68 | 1.2g | m.p. 85.5-86.5° C |
| 2-bromo-7-methoxy-1-tetralone, 6.5 (25.5 mmol) | 7-methoxy-1-naphthol76 | 3.8g | M.p. 103-104° C |
6-methoxy-1-naphthoyltriflate 69:
To a solution of 2.3g (13.21 mmol) of 6-methoxy-1-naphthol in 50mL of pyridine, 2.6mL (15 mmol) of trifluoromethansulfonic anhydride was added dropwise at 0° C. The solution was warmed to room temperature and stirred overnight, then quenced with water, and extracted with ether. The combined extracts were washed with 10% HCl until the aqueous solution was acidic. The solution was dried (MgSO4), and the solvent was removed in vacuo. The residue was chromatographed (petroleum ether/ethyl acetate 12:1) to give 3.4g (84%) of product as a yellow oil.
| Naphthol | Product | Yield | Appearance |
| 6-methoxy-1-naphthol, 2.3g (13.21 mmol) | 6-methoxy-1-naphthoyltriflate 69 | 3.4g (84%) | yellow oil |
| 7-methoxy-1-naphthol 4.0g (26.8 mmol) | 7-methoxy-1-naphthoyltriflate 77 | 4.9g (70%) | Yellow oil |
6-methoxy-1-naphthoic acid 70:
To a solution of 0.96g (3.18 mmol) of 6-methoxy-1-naphthoyl triflate (69) in 30mL of DMF, was added 34mg (0.15 mmol) of palladium (II) acetate, followed by 40mg (0.10 mmol) of 1,3-bis(diphenylphosphino)-propane, and 1.20mL (8.6 mmol) of triethylamine. The mixture was flushed with carbon monoxide for 25 min, 3.1mL of 96% formic acid was added dropwise and the reaction was stirred for 6 h under a carbon monoxide atmosphere at room temperature. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic extracts were washed with five portions of brine, followed by two portions of saturated aqueous NaHCO3. The aqueous bicarbonate extracts were combined, neutralized with 10% HCl and again extracted with ether. The ethereal extracts were washed with water and dried (MgSO4); the solvent was removed in vacuo to give 0.42g (65%) of crude product as a yellow powder. Recrystallization (ethyl acetate/petroleum ether 1:1) gave 0.35g of yellow needles: m.p. 185-186° C
| Triflate | Product | Yield | M.p./appearance |
| 6-methoxy-1-naphthoyl triflate, 0.96g (3.18 mmol) | 6-methoxy-1-naphthoic acid 70 | crude: 0.42g (65%)/recrystallized: 0.35g | m.p. 185-186° C/yellow needles |
| 7-methoxy-1-naphthoyl triflate, 1.06g (3.5 mmol) | 7-methoxy-1-naphthoic acid 77 | crude:0.50g (70%)/recrystallized: 0.42g | m.p. 170-172° C/yellow needles |
2-Methyl-1-pentyl-3-(6-methoxy-1-naphtho
36:
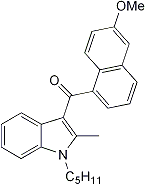
To a solution of 0.20g (1.0 mmol) of 6-methoxy-1-naphthoic acid in 10mL of dichloromethane, 0.44mL (5.0 mmol) of oxalyl chloride was added dropwise. The resulting solution was stirred at room temperature for 1 h, then refluxed for 1 h. After cooling, the solvent and residual oxalyl chloride were removed in vacuo. The solid residue was dissolved in 5mL of toluene, and added to a solution of 0.24g (1.2 mmol) of 2-methyl-1-pentylindole in 4mL of toluene at 0° C. To this solution was added dropwise 0.82mL (1.8M, 1.5 mmol) of ethylaluminum dichloride. The mixture was allowed to warm to room temperature and stirred for 18 h. After quenching with water, the reaction mixture was extracted with ether and the ethereal extracts were washed with water and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed (petroleum ether/ethyl acetate 12:1) to give 0.23g (60%) of product: m.p. 114.5-116° C.
| Naphthoic acid | Indole | Product | chromatographed | Yield | m.p. or appearance |
| 6-methoxy-1-naphthoic acid 0.20g (1.0 mmol) | 2-methyl-1-pentylindole 0.24g (1.2 mmol) | 2-Methyl-1-pentyl-3-(6-methoxy-1-naphtho |
p.e/e.a 12:1 | 0.23g (60%) | 114.5-116° C |
| 6-methoxy-1-naphthoic acid 0.19g (0.9 mmol) | 2-methyl-1-propylindole 0.21g (1.3 mmol) | 2-methyl-1-propyl-3-(6-methoxy-1-naphtho |
p.e./e.a 8:1 | 0.19g (56%) | green oil |
| 6-methoxy-1-naphthoic acid 0.19g (0.9 mmol) | 1-pentylindole 0.24g (1.3 mmol) | 1-Pentyl-3-(6-methoxy-1-naphthoyl)indole |
p.e./e.a 8:1 | 0.29g (81%) | green oil |
| 6-methoxy-1-naphthoic acid 0.18g (0.9 mmol) | 1-propylindole 0.20g (1.3 mmol) | 1-Propyl-3-(3-methoxy-1-naphthoyl)indole |
p.e./e.a 8:1 | 0.19 (55%) | colorless crystals m.p. 139-140° C |
| 7-methoxy-1-naphthoic acid, 0.18g (0.9 mmol) | 2-methyl-1-pentylindole, 0.24g (1.2 mmol) | 2-methyl-1-pentyl-3-(7-methoxy-1-naphtho |
p.e./e.a. 8:1 | 0.19g (43%) | viscous oil |
| 7-methoxy-1-naphthoic acid, 0.18g (0.9 mmol) | 2-methyl-1-propylindole, 0.20g (1.2 mmol) | 2-methyl-1-propyl-3-(7-methoxy-1-naphtho |
p.e./e.a. 8:1 | 0.20g (65%) | viscous oil |
| 7-methoxy-1-naphthoic acid, 0.19g (1.0 mmol) | 1-pentyindole, 0.24g (1.2 mmol) | 1-pentyl-3-(7-methoxy-1-naphthoyl)indole |
p.e./e.a. 8:1 | 0.21g (66%) | viscous oil |
| 7-methoxy-1-naphthoic acid, 0.18g (0.9 mmol) | 1-propylindole, 0.24g (1.2 mmol) | 1-propyl-3-(7-methoxy-1-naphthoyl)indole |
p.e./e.a. 8:1 | 0.19g (64%) | viscous oil |
[(ChemisTris note) = the original entries for 37, 39 and 41 state that R-1-pentylindole (rather than R-1-propylindole) is used to produce the R-1-propyl-3-([6- or 7-]methoxy-1-naphthoyl)indole. This is one of several errors in the dissertaion - it seems like the author was a bit careless with the write-up (I'm guessing that the mistakes are artifacts of cutting and pasting, after all it's ~150 pages) - no doubt I have not caught all the errors.
Selected refs for cannabimimetic indoles:
[29] Huffman, J.W.; Dai, D.; Martine, B.R.; Compton, B.R. Bioord. Med. Chem. Lett. 1994, 4(4), 563
PDF: http://mishmashblue.tripod.com/29.pdf
[30] Wiley, J.L; Compton, D.R; Dai, D.; Lainton, J.A.H.; Phillips, M.; Huffman, J.W.; Marin, B.R. J. Pharmacol. Exp. Ther. 1998, 285, 995.
PDF: http://mishmashblue.tripod.com/30.pdf
[31] Razdan, R.K. Pharmacol. Rev. 1986, 38[/], 75.
[41] Song, Z.H.; Bonner, T.I. [i]Mol. Pharmacol. 1996, 49, 891
[42] Chin, C.; Lucas-Lenard, J.; Abadji, V.; Kendall, D.A. J. Neurochem 1998,70(1), 366
PDF: http://mishmashblue.tripod.com/42.pdf (there was an error with the pdf thumbnails (from the journal server end?)
[43] Bramblett, R.D.; Panu, A.M.; Ballesteros, J.A.; Reggio, P.H. Life Sci. 1995, 56(23-24) 1971
PDF: http://mishmashblue.tripod.com/43.pdf
[44] Reggio, P.H.; Basu-Dutt, S.; Barnett-Noris, J.; Castro, M.T.; Hurst, D.P.; Seltzman, H.H.; Roche, M.J.; Gilliam, A.F.; Thomas, B.F.; Stevenson, L.A.; Pertwee, R.G.l Abood. M.E. J. Med. Chem 1998 41(26), 5177.
PDF: http://mishmashblue.tripod.com/44.pdf
DOI:10.1021/jm9801197
[45] Huffman, J.W.; Wu, M.J.; Lainton, J.A.H.; Dai, D.; Phillips, M.; Keel, C.; Wiley, J.L.; Compton,D.R., Showalter, C.; Abood, M.E.; Martin, B.R.; Symposium on the cannabinoids, international Cannabinoid Research Society; Burlington, VT, 1997 pp 8.
[50] Augustine, R.L.; Gustaven, A.J.; Wanat, S.F.; Pattison, I.C.; Houghton, K.S.; Koletar, G. J. Org. Chem 1973, 38(23), 4073
PDF: http://mishmashblue.tripod.com/50.pdf
Got democracy?http://www.dhushara.com/book/multinet/de