04-28-02 01:23
No 302465
When doing a frac vac distill on mdp2p does a distillation temperature range of 110 to 155 sound reasonable?
It probably would be because the distillate starts coming over at 110 and never stops coming over until the temp starts to back off from 155 and the distillate never changed appearance throughout that temp. range.
Pale slightly orange yellow with a hint of green.
(Hive Bee)
04-28-02 15:31
No 302724
The pump is a Bestech SV series Rotary Vane Vacuum Pump.
2 Stage direct driven. It's specs say its ultimate vacuum is .01 torr but I sure don't see that. Not even close to that. If my calculations are correct the pump is pulling 30 torr but there may be a leak and I will have to check that out my completely.
This is the 3rd vac frac distill on this stuff. The first two were complete failures but the 3rd was really well behaved and proceded pretty much as a good vac frac distill should.
The stuff does not smell like sassy oil except for very very faintly.
The article by Methyl Man indicates that a small amount of safrole will not affect the Hg/Al Amalgam rxn.
One would expect the temp to fall once the safrol had been brought over and then a short time later the temp would start to rise again as the higher boiling point component came over. What happened was that the temp never did fall but kept rising steadily through that range with a constant drip drip drip of the distiallate. So at what point would you back off the vac and put a new receiving flask on?
Usually once the temp had fallen then you would change the flask.
I really don't see the point of a 4th vac frac distill on this stuff because the behavior would be the same.
I suspect that this material will be processed through the Al/Hg rxn in the hopes that the bulk of what there is will be the desired ketone. The Hg/Al rxn is specific for ketones. Perhaps you have some ideas on how to clean up this 43 grams of distillate but how many vac frac distills can you subject this ketone to anyway before it just degrades itself out of existence?
(Chief Bee)
04-28-02 17:21
No 302769
Degradation-wise, you can distill it a whole lot of times before it degrades, but the mechanical losses in fractional distillations are worth noting, especially if you are using a larger setup than 100ml/NS14 for that quantity.
(Hive Bee)
04-28-02 18:35
No 302793
Is the distillate coming over too fast? Is your SWIM heating the flask too fast? What type of column is used?
The reflux ratio = drops of liquid falling back from the column divided by drops of distillate recovered. It, supposedly, is supposed to meet or exceed the # of theoretical plates in your column. For instance, if you have an 80cm Vigreux with 2.5 theoretical plates, you should see at least 2-3 drops fall back into the distilling flask for every drop of distillate recovered.
SWIM saw once on Mr Wizard-- A batch was being distilled.. His hot plate was lousy and took FOREVER to get up to temperature.. The first distillate collected was some alkene. The temperature drops, and then raises again as the product distilled over.
Julia
(Hive Bee)
04-28-02 19:44
No 302815
Well, to separate the safrole from the mdp2p we would require 5 theoretical plates which can be obtained by using a 20 cm glass column packed with glass helices. A corresponding 20 cm Vigreux column would give you 2.5 TP's.
With this in mind I can see that my frac column was too small and had too few TP's. This would translate into poor resolving power for the column used and the compounds involved.
The big problem with the larger frac columns is the increased hold-up that stays in the column and this becomes significant when you are working with small batches.
It's always a tradeoff.
(Hive Bee)
04-28-02 20:29
No 302830
I reckon your pulling 3 to 4 mbar to get that range for MDP2P. The temp range of 130-140 deg at that vac would be your goal. Any higher 140 ish to 150s and you would notice as the vac is removed the color of the higher boiling distillate turns a really deep orange straight away and smells very different form the rest of lower bp fraction. SWIM would just collect the 110-140 ish fraction and use that, since the safrole/isosafrole won't interfere in the reductive amination step but any ketone that was distilling over with it will.
Try to get the feel for the distillation rather than strictly following the temps. Especially if you are not measuring your vac. Since the tiniest little vac leak can set you off on the wrong direction. Less viscous with a lighter color is your azeotrope, the stronger fluro color and change in viscosity is your main bulk of MDP2P and then more viscous again and orangey colored distillate means its time to pull the plug.
(Hive Bee)
04-28-02 22:36
No 302879
Thanks for all that great information which I will be sure to keep in mind next time I give it a try. A vacuum gauge would be nice and make life a lot easier with this stuff. As usual its the $$$. Four mbar translates into 3 torr but it appears as this pump is pulling more like a dismal 30 torr. With a vacuum gauge I would know for sure. The gauge is looking like a must-have item for this kind of work.
(Newbee)
04-29-02 08:34
No 303005
Let me clear something up. An azeotrope is not a mixture that produces distillate containing both components with a boiling point in between the two pure component boiling points. Azeotropes are a minimun or maximum on the bubble-point/composition curve in which the vapor and liquid have the same composition. I won't get into the advanced thermodynamics, but basically, an azeotrope either boils below the more volatile component (miniumum azeotrope) or above the less volatile component (maximum azeotrope). Furthermore, minimum azeotropes are much more common. If safrole and ketone formed an azeotrope under 10 mm vacuum, it would either be below 105C (safrole bp) or above 145C (ketone bp).
DiMethyl: when you run an inefficient distillation (less than two T.P.'s of seperation) the whole range of distillate will have a yellow tint, although it will become more yellow and viscous as the temperature increases. This is because a 50/50 mol% mixture of safrole/ketone will first boil at around 120C and produce vapor containing about 30% ketone/70% safrole (These numbers are just estimates). This is what simple distillation will give you for the first drop of distillate (no column). As you continue to distill, more safrole vaporizes than ketone, so the solution in the boiling flask will shift to a higher ketone fraction over time. As the mole fraction of ketone becomes higher in the boiling flask, the boiling point of the solution also rises. Now, the temperature on the thermometer in the distillation head is the dew point of the vapor, which is the same as the boiling point of the solution, but higher than the boiling point of the distillate (for simple distillation only).
Fractional distillation is completely different. Simple distillation represents 1 T.P. of seperation. Now, suppose you have a column that gives you 2 T.P. of separation. Add 1 T.P. to it due to the simple distillation of the boiling solution to the bottom of the column, and you get 3 T.P. of total separation. This means that for every drop of distillate collected, the separation is equivalent to boiling an infinitely large amount of solution in the flask once and collecting the distillate (so that the composition is constant). Then taking an infinitely large amount of distillate and running another simple distillation on it. Then repeat this one more time on the second distillate to obtain distillate of the same composition that you get by using the column on an infinitely large amount of solution.
It gets more complicated. Remember how we assumed that the column gave 2 T.P. of seperation? Well that is only true is the rate that distillate is collected is infinitely low, or the reflux ratio is very large (reflux ratio is the rate of liquid returning to the flask divided by the rate of vapor ascending the column. In other words, the mass flow of vapor up the column divided by the mass flow of liquid down the column). In other words, if you stuck a vertical condenser on top of your column, this is the composition of the liquid driping off the condenser. When ever you begin removing distillate, you lower the efficiency of the column. If the reflux ratio is 0 (All of the vapors are collected as distillate). Your column efficiency is 0 and you are in effect doing a simple distillation. In expensive distillation head, there is a valve you can adjust to controll the rate of removal of distillate. Unfortunately, the common aparatus does not have this feature. All of the liquid flowing down the column is condensed by heat loss through the column wall and distillation head. This rate of condensation is more or less fixed, no matter how fast you distill. You do have control over the rate of distillate produced, however. You can't really measure the rate of liquid flowing down the column by counting the drops. Most of it streams down the walls. Something you could try to give you a better feel of this concept is to start distilling your mixture at a slow rate Then jack up the hotplate and watch the temperature at the head climb. Now lower the oil bath temperature and watch the head temperature fall again. You pretty much have a balance here -- time it takes to distill vs. efficiency of the separation. That is why everyone stresses to distill slowly.
I know it's all pretty complicated, but if you want to understand it better, study thermodynamics. It's a very interesting subject.
(Hive Bee)
04-29-02 11:12
No 303057
Thanks for taking the time to share all that information with us. It looks like the vacuum fractional distills are the key to being successful with this kind of work. In comparison the rest of it seems easy. I guess it all boils down to using enough TP's and doing the distillation as slowly as you can. In my case there were not enough TP's so that although I was only collecting a drop every 2 to 3 seconds, the seperation was not occuring properly. Big columns and lots of time.
(Daddy)
04-29-02 20:41
No 303223
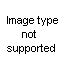
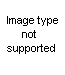
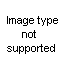
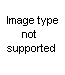
The Oldershaw distilling columns include such advantages as speed in reaching equilibrium, high reflux rate, low operating holdup, uniformity of operation and all-glass construction.
The column consists of a series of sealed-in perforated plates, each with a drain which directs the condensed distillate to the opposite side of the next lower plate and a weir in each plate to maintain a uniform liquid level. Ascending vapors pass through the perforations to form uniform bubbles in the liquid flowing across each plate. All holes are drilled, ground and checked for uniformity to insure proper operation.
http://www.lab-glass.com/html/nf/DSTA-LG
http://www.lab-glass.com/html/nf/DSTA-LG
http://www.lab-glass.com/html/nf/DSTA-LG
http://www.lab-glass.com/html/nf/DSTA-LG
LT/
WISDOMwillWIN
(Newbee)
04-30-02 14:32
No 303481
I got one of these. They kick ass! Combined with my reflux controller, I can separate just about anything.
(Chief Bee)
04-30-02 15:13
No 303504
What is a reflux controller?
(Stoni's sexual toy)
04-30-02 16:21
No 303517
Device which lets you select the distillation to reflux ratio. E.g., if you have way too much time on your hands you can distill stuff with a reflux ratio of 1:200, meaning for every drop reaching your receiving flask 200 drops will fall back into the column.
I'm not fat just horizontally disproportionate.
(Newbee)
04-30-02 16:32
No 303523
http://www.lab-glass.com/html/nf/DSTA-LG
With this bitch, you can distill shit fast and pure. Takes only about two to three hours to distill 1.5L of safrole with a 5�C temperature range.
(Stoni's sexual toy)
05-02-02 00:40
No 304114
Using such a thing for safrole isolation and then speeding the distillation that much is counterproductive. Column efficiency is best with low throughput, and most columns will be flooded using such distillation rates. Flooded colums have a very shitty separation efficiency. Another problem with columns is that many types aren't suited for vacuum distillation since the pressure on top of the column is lower than the pressure inside the boiling flask.
If you push 1 liter of sassy through a 2feet column per hour and use such a column head thingy then you could achieve the same separation efficiency with a much cheaper distillation unit since you are operating way outside its rating.
Why do people always rush it? Time is money I know, but such a thing can be left running overnight, or you can do something else while the distillation unit is running. They do not require 100% attention.
I'm not fat just horizontally disproportionate.
(Chief Bee)
05-02-02 01:16
No 304125
Where can I read about the function of all the stranger ground-glass equipment that is sold from labglass/chemglass/aldrich etc? They often look nice, costs a fortune, but I still have no idea about the possible uses and advantages...
(Stoni's sexual toy)
05-02-02 02:55
No 304138
HOH!
Read again what I wrote!
if you want to distill 1L of sassy per hour then distill it without column, because the separation efficiency would be nearly the same. It makes no sense to distill sassy or other stuff at such a rate through regular lab sized equipment!
If you want to distill 1 liter of sassy per hour without losing separation efficiency then you need a column with 10cm/4inches internal diameter.
You guys have to learn how to multitask, I can do 2 or three reactions at the same time, so quit telling us you are currently processing 1L of sassy per hour and sassy distillation is the step that is limiting your daily output.
Rhodium: post pics and I'm sure someone will be able to tell you what the purpose of the glass is.
I'm not fat just horizontally disproportionate.
(Hive Bee)
05-02-02 12:44
No 304281
I had the same problem rhodium... what I found was the lab technician books told you how to set them up for the experiments, and the name of the apparatus in the catalogs was a good key on where to look in the index.
Infinite Radiant Light - THKRA
(Chief Bee)
05-02-02 18:05
No 304395
Do you have any lab technique book suggestions besides Vogel's? As I have no idea about what some glassware is good for, or two to me equivalent distillation setups may have widely differing separation efficience, I don't know where to start to learn about all the existing glass apparatuses (apparati?)... Also, even if I would buy a $3500 glass monster from aldrich, there is still no manual how to operate the thing in the most effective fashion.
(Stoni's sexual toy)
05-03-02 02:25
No 304544
Aldrich also sells books, I'm sure they have lots of it covered in there. But I would never buy anything from fucking Aldrich. Their glass prices are obscene! Sure, they have a lot of stuff (lots of it pretty unnecessary), but I can get most of it for a fraction of the money locally. Also, yesterday night I checked some chemicals prices. These fuckers buy stuff produced and sold by the industry for a few pennies and then fucking sell it at 20 times the price! I am not kidding, I have ordered stuff from the original manufacturer and from Aldrich, and they were EXACTLY the same except the price difference.
Edit: They also raised prices by 10% this year even for the most basic stuff.
Ooops, got a little carried away.
Rhodium, if you want to learn e.g. about different distillation apparatuses etc. then go to your local library, I'm sure they have several books about distillation techniques. Lots of it can also be figured out once you know the basic concepts and use some common sense.
I've never seen a book solely about glassware and its uses.
I'm not fat just horizontally disproportionate.
(Hive Bee)
05-03-02 02:38
No 304546
I think that the best practical info is in the chemistry book's from the 50 and 60�s. By the way, wacumjaced collums are greth. I have a wacum jacked Vigreux collum, an it beats all my other collums (packed and non packed, but no wacumjacked). I don't think they are that ekspensive.
(Newbee)
05-03-02 07:40
No 304583
It's a bubble plat column. These are not as easily flooded as vigreaux columns are. The trick to getting pure, fast safrole, is to vary your distillation rate throughout the process. The beginning and end are crucial for purity, so you must go slow during these parts. During the middle, however, you can crank it up full blast and it won't affect your purity one bit. This is how I do it
1) Distill very slowly until the temp is 3C below your expected bp.
2) Distill very fast until the temp goes 2C above your expected bp.
3) Now distill very slowly until the temp goes 3C above your expected bp.
This way, you don't wasted time one the middle shit, just the two ends.
(Hive Addict)
05-03-02 09:43
No 304604
There's only two factors in separation efficiency:
1) reflux ratio (how many drops fall back into the flask versus how many drops jump over into the receiving flask)
2) theoretical/actual plates
If the reflux ratio is extremely high (ie operating at total reflux), then the separation is as good as the theoretical plates indicate. If less, then the operating line steps off from the y=x line on the macabe-thiele diagram (vapor pressure of light component vs vapor pressure of heavy component). And of course, the reflux ratio is controlled by the heat loss on the column. For something like ethanol/water you want to remove a lot of heat, for something like safrole boiling at 200C you want to remove almost no heat.
(Stoni's sexual toy)
05-03-02 15:56
No 304691
Another important factor is the amount of stuff going in and out of your column. The less the better from a separation efficiency standpoint.
I'm not fat just horizontally disproportionate.
(Hive Bee)
05-04-02 02:33
No 304833
I thought we were not supposed to list sources? But now that the cat is out of bag I have a number of pieces of glassware with labglass printed on them. I think they come from Mexico. Works great for me. The Oldershaw column is going on my wish list.
(Daddy)
05-04-02 02:33
No 304834
Keyword: PATIENCE!
Simple logic: you choose the length of the vigreux column on a few data:
1. What are you trying to reflux, a high or low boiling substance.
2. Is your column insulated or not, and is it needed at all. High=yes , Low boiling=not
2a. Choose the best insulation material for high boiling stuff, the better the insulation, the more preciser your reflux separation in f.ex. a 1 to 5 Celsius reflux-traject.
3. If you have f.ex. 3 vigreux columns, 10, 20 and 40 cm, does the 40 cm allow you to receive sufficient flowrate in your receiving flask? If yes, use that one, if no, step down untill you have found the right one.
If you want to know really what's needed for calculating these things, which you can easily do by trial and error, then you have to read sticky threads and UTFSE.
And buy an Oldershaw column + the rest. LT/
WISDOMwillWIN
(Hive Addict)
05-04-02 14:03
No 304959
If it's a boiling at a low temp w/high flow rate, say distilling multiple liters of ethanol at 78C... you might actually want a large column with external cooling. Take a look at: ../rhodium/archive/still.pdf
If you're boiling at a medium temp or a low temp w/a low flow rate... you'd just want the column (eg distilling acetone, DCM, etc to reprocess them... not to be confused with stripping solvents, where you likely want no column...). Or perhaps if all the vapors are coming back as reflux, (say distilling PERC at ~120C with a 400mm column) you might want insulation (say, some pieces of fleece... or a vacuum jacketed column).
If you're boiling at a high temp with a low flow rate, you want VERY little cooling. Or else all of it will be returned as reflux, so you need good insulation. Say you're distilling safrole at 180C over a 200mm column. You want to wrap the column with something that insulates well and can withstand the temperature, say wrapping glasswool around your column. Alternatively, you can get a vacuum jacketed column.
Also, it should be noted that the type of packing is important too. Vigreux's acheive very few HETP compared to say steel wool in the same diameter/length. Although stainless steel wool is really good at doing its job... it won't stand up to all conditions (and has a higher hold up)... and cleaning has an influence on what column you use as well.
I hope that hasn't confused people. Basically you want enough insulation (occassionally cooling or heating) so you get a decent reflux ratio and that the column is efficient enough (length, diameter, and choice of packing) to see a clean separation between fractions.
(Hive Addict)
05-04-02 17:59
No 305033
I'm a little confused as to why you would add stainless steel wool if you're getting too much reflux. A quick review:
Stainless steel wool is a packing material for the column. It's put inside of a hollow column. It's best suited for small diameter columns (<3") where raschig rings and other materials and glass indentations (such as on a vigreux) aren't efficient enough for the purposes desired. Raschig rings are used for medium sized columns... industrial-sized columns use stages with plates. Btw, you won't be able to get stainless steel wool to work with a vigreux (the indentations stop you from putting it thru) but you could use it with a liebig condenser (when the liebig condenser is used vertically as a column... not as a condenser... I hope that's was obvious).
Glass wool is for insulating the column if you're getting to much reflux. It's wrapped around the outside of the column. (most hardware stores carry it for wrapping pipes) Fleece is much easier to handle and I've found it to insulate even better to insulate than glasswool if you're distilling at a temperature where the fleece won't melt. Cotton is a really poor insulator, but works for higher temps as well.
Crumpled aluminum foil works some, but any of the other techniques is highly recommended.
Btw, if you only need to insulate part of your column to acheive a good reflux ratio, ensure it's the bottom part that is insulated.