(Chief Bee)
01-16-04 09:38
No 482763
(Rated as: excellent)
Ueber den alpha-Phenylacetessigester
Walter Beckh
Chem. Ber. 31, 3160-3164 (1898) (../rhodium/pdf /p2p.phenylac
Summary:
Sodium ethoxide catalyzed condensation of ethyl acetate with phenylacetonitrile, forming alpha-Phenylacetoacetonitrile, which upon treatment with sulfuric acid is simultaneously hydrolyzed to the acid and decarboxylated to give phenylacetone.
____ ___ __ _
Ueber Phenacetylmalonsäureester
Hermann Metzner
Ann. 298, 374-378 (1897) (../rhodium/pdf /p2p.phenylac
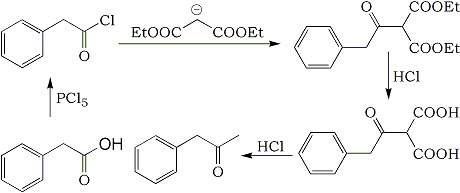
Summary:
Phenylacetic acid is chlorinated with PCl5 in chloroform, yielding phenylacetyl chloride in 88% yield. This is then used to acylate sodium diethylmalonate to give the phenylacetylmalonic ester in 75% yield by leaving the mixture undisturbed for a week. Refluxing the phenylacetylmalonic ester in 20% (1.1 g/mL) aqueous hydrochloric acid for a few hours results in double ester hydrolysis and decarboxylation, leaving phenylacetone as the end product.
____ ___ __ _
Oxydationsprodukte der Benzylketone
A. Popoff
Chem. Ber. 5, 500-502 (1872) (../rhodium/pdf /p2p.me2zn.pd
Summary:
Alkylation of phenylacetyl chloride with dimethyl zinc under cooling, followed by hydrolysis of the intermediate with dilute hydrochloric acid gave crude P2P, bp 210-217°C. After purification via the crystalline bisulfite adduct, the ketone had bp 214-216°C.
____ ___ __ _
Zur Geschichte der Phenylessigsäure
B. Radziszewski
Chem. Ber. 3, 198-199 (1870) (../rhodium/pdf /p2p.paa-ba-a
Summary
Phenylacetone is prepared by pyrolysis of phenylacetic acid and barium acetate (1:1, w/w). The distillate also contains acetone, toluene and diphenylacetone. The phenylacetone is then isolated by fractional distillation, bp 215°C, d. 1.010 g/mL at 3°C.
The Hive - Clandestine Chemists Without Borders
(Chief Bee)
01-21-04 00:32
No 483632
Monooxygenase activity of cytochrome c peroxidase
VP Miller, GD DePillis, JC Ferrer, AG Mauk and PR Ortiz de Montellano
J. Biol. Chem. 267, 8936-8942 (1992) (http://www.jbc.org/cgi/reprint/267/13/8
Abstract
Recombinant cytochrome c peroxidase (CcP) and a W51A mutant of CcP, in contrast to other classical peroxidases, react with phenylhydrazine to give sigma-bonded phenyl-iron complexes. The conclusion that the heme iron is accessible to substrates is supported by the observation that CcP and W51A CcP oxidize thioanisole to the racemic sulfoxide with quantitative incorporation of oxygen from H2O2. Definitive evidence for an open active site is provided by stereoselective epoxidation by both enzymes of styrene, cis-beta-methylstyrene, and trans-beta- methylstyrene. trans-beta-methylstyrene yields exclusively the trans- epoxide, but styrene yields the epoxide and phenylacetaldehyde, and cis- beta-methylstyrene yields both the cis- and trans-epoxides and 1-phenyl- 2-propanone. The sulfoxide, stereoretentive epoxides, and 1-phenyl-2- propanone are formed by ferryl oxygen transfer mechanisms because their oxygen atom derives from H2O2. In contrast, the oxygen in the trans- epoxide from the cis-olefin derives primarily from molecular oxygen and is probably introduced by a protein cooxidation mechanism. cis-[1,2-2H]- 1-Phenyl-1-propene is oxidized to [1,1-2H]-1-phenyl-2-propanone without a detectable isotope effect on the epoxide:ketone product ratio. The phenyl-iron complex is not formed and substrate oxidation is not observed when the prosthetic group is replaced by delta-meso-ethylheme. CcP thus has a sufficiently open active site to form a phenyl-iron complex, to oxidize thioanisole to the sulfoxide, and to epoxidize styrene and beta-methylstyrene.
The Hive - Clandestine Chemists Without Borders
(Chief Bee)
01-26-04 06:03
No 484608
(Rated as: excellent)
Aerogel Catalysts
Thoria: Preparation of Catalyst and Conversion of Organic Acids to Ketones
S. Swann, E. G. Appel, S. S. Kistler
Ind. Eng. Chem. 26(4), 388-391 (1934) (../rhodium/pdf /p2p.thoria-1
Abstract
The conversion of aliphatic Acids to ketones has been studied over thoria aerogel. It has been found that the aerogel catalyst is distinctly superior to thoria hydrogel, thoria prepared from the oxalate, and thoria on pumice for this purpose. The yields of ketones compare favorably with the best reported in the chemical literature.
____ ___ __ _
Thoria Aerogel Catalyst: Aliphatic Esters to Ketones
S. Swann, E. G. Appel, S. S. Kistler
Ind. Eng. Chem. 26(9), 1014 (1934) (../rhodium/pdf /p2p.thoria-1
The Hive - Clandestine Chemists Without Borders
(Chief Bee)
04-25-04 01:02
No 502749
(Rated as: excellent)
Alkyl Benzyl Ketones and Hydantoin Derivatives
E.H. Sund and H.R. Henze
Journal of Chemical and Engineering Data 15(1), 200-201 (1970)
Ten alkyl benzyl ketones were synthesized by the interaction of phenylacetyl chloride and the requisite dialkyl cadmium, the synthesis being modeled after a published procedure by Blaise [Compt. Rend. 133, 1218 (1901)].
Experimental
Preparation of Phenyl-2-Propanone
A mixture of 40 ml. of anhydrous ether and 6.1 grams (0.25 mole) of magnesium was stirred under reflux while 35.5 grams (0.25 mole) of methyl iodide in 140 ml. of anhydrous ether was added over a 3-hour period; stirring under reflux was continued for an additional hour. The reaction mixture was cooled with an ice bath and 22.4 grams (0.134 mole) of powdered anhydrous cadmium chloride was added over a 5- to 10-minute period, warmed to room temperature, and refluxed on a steam cone for 1 hour. Ether was removed by distillation on a steam bath. To theresidue was added 100 ml. of anhydrous benzene and the distillation was continued until about 50 ml. more of distillate was collected. Again 100 ml of anhydrous benzene was added, the flask was cooled in an ice bath, and 30.9 grams (0.2 mole) of phenylacetyl chloride in 75 ml of anhydrous benzene was added with stirring over a period of approximately 10 minutes. The reaction mixture was warmed to room temperature and refluxed with stirring on a steam cone for 1 hour. The flask was again cooled in an ice bath and the reaction mixture decomposed by the addition of a solution of 25 grams of ammonium chloride in 200 ml of cold water. The organic phase was separated, washed, and dried over anhydrous sodium sulfate. The benzene was removed by flash distillation and the ketone distilled under reduced pressure. There was thus obtained 15.5 grams (58%) of 1-phenyl-2-propanone, bp 74-76°C/3 mmHg).
The Hive - Clandestine Chemists Without Borders
(Chief Bee)
04-26-04 18:32
No 503140
(Rated as: excellent)
Synthesis of Methyl Ketones from Diethyl Acylmalonates
Howard G. Walker and Charles R. Hauser
J. Am. Chem. Soc. 68, 1386-1388 (1946) (../rhodium/pdf /p2p.diethyla
A convenient method for preparing certain methyl ketones consists in the acylation of the sodium or, preferably, the magnesium-ethoxy derivative of diethyl malonate with the appropriate acid chloride, followed by hydrolysis and decarboxylation of the two ester groups of the resulting diethyl acylmalonate in the presence of acid, thus:
In the present investigation, satisfactory yields [was obtained of] phenylacetone1. We have chosen the magnesium ethoxy derivative of diethyl malonate2 rather than the sodium derivative because we believe the former is prepared more conveniently. Although the acid chloride does not appear to react appreciably with the excess alcohol2 used in the preparation of the magnesium-ethoxy derivative, we have employed a 10% excess of the latter in order to minimize this possible side reaction. The crude diethyl acylmalonates were hydrolyzed and decarboxylated in the presence of aqueous acetic and sulfuric acids according to the method previously employed for the ketonic cleavage of certain beta-keto esters3. The present method appears to be one of the best for the preparation of certain higher aliphatic or aliphatic-aromatic4 methyl ketones [...].
Experimental
Diethyl acylmalonates were prepared by a modification of the procedure of Lund2,5
In a 500-ml three-necked flask equipped with a mercury-sealed stirrer, dropping funnel, and reflux condenser protected by a drying tube, was placed 5.35 g. (0.22 mole) of magnesium. Five ml of absolute ethanol and 0.5 ml of carbon tetrachloride were added. The reaction, which started almost immediately, was allowed to proceed for a few minutes and 75 ml of absolute ether was then added cautiously. The resulting mixture was placed on the steam bath and a solution of 35.2g (0.22 mole) of diethyl malonate, 20 ml of absolute ethanol and 25 ml of absolute ether was added at such a rate that rapid refluxing was maintained, heat being applied when necessary. The mixture was refluxed for three hours, or until the magnesium had dissolved. To the clear solution was added with vigorous stirring6 an etheral solution of 30.8g (0.20 mole) phenylacetyl chloride and the mixture refluxed for one-half hour. The reaction mixture was cooled and acidified with dilute sulfuric acid. The ether phase, with which an ether extract of the aqueous phase was combined, was washed with water and the solvent distilled.
To the crude diethyl phenylacetylmalonate was added a solution of 60 ml of glacial acetic acid, 7.5 ml of concentrated sulfuric acid and 40 ml of water, and the mixture refluxed for four or five hours until the decarboxylation was complete. The reaction mixture was chilled in an ice-bath, made alkaline with 20% sodium hydroxide solution, and extracted with several portions of ether. The combined ethereal extracts were washed with water, dried with sodium sulfate followed by Drierite, and the solvent distilled. The residue containing the ketone was distilled in vacuo to give Phenyl-2-Propanone in 71% yield (bp 97-98.5°C/13 mmHg, 214-215°C/760mmHg).
References
[1] Metzner prepared phenylacetone by this method, but no yield was reported: Ann. Chem. 298, 378 (1897) (../rhodium/pdf /p2p.phenylac
[2] Lund, Chem. Ber. 67B, 935 (1934)
[3] Hudson and Hauser, J. Am. Chem. Soc. 63, 3163 (1941)
[4] We believe that the malonic ester method is more convenient for phenylacetone than those described in Org. Synth. Coll. Vol. II, 389, 391 (1943) (http://www.orgsyn.org/orgsyn/prep.asp?p
[5] The present procedure is similar to that of Breslow, Baumgarten, and Hauser [J. Am. Chem. Soc. 66, 1286 (1944)] for the preparation of ethyl tert-butyl acylmalonates.
[6] In certain cases a viscous mixture was formed and unless it was stirred vigorously, lower yields were obtained.
The Hive - Clandestine Chemists Without Borders
(Stranger)
08-30-04 18:32
No 528275
Wow this is great, I'm really liking the Pd catalyzed alpha arylation and the bromobenzene with propylene oxide to P2Pol and then oxidized to P2P. Several questions though, is 1-Phenyl-2-nitropropene a regulated precursor? I didn't find it in the DEA handbook although all of its precursors like benzaldehyde and nitroethane were regulated. Secondly, I assume the procedure with calcium acetate can be done directly with phenyl acetic acid not its calcium salt, no? Also does anybody else think some of these are a bit impractical except as a possible laboratory curiosity? I saw a couple of procedures where the precursors aren't even commercially available, not regulated, it's just that the lab companies simply don't have them.
Trogdor was a man. A dragon man. Or maybe just a dragon...
08-30-04 20:32
(Rated as: Check your facts + Leave the moderation to the moderators)
(Chief Bee)
09-01-04 21:49
No 528916
is 1-Phenyl-2-nitropropene a regulated precursor? I didn't find it in the DEA handbook although all of its precursors like benzaldehyde and nitroethane were regulated.
It is not explicitly regulated on a national level as far as I know*, but due to the noxious and unstable nature of nitroalkenes (not to mention a rather low licit demand), they are seldom offered for sale from chem suppliers. But even if the sale of 1-Phenyl-2-nitropropene isn't explicitly prohibited, it sure is a suspicious item and commercial aquisition should be avoided.
Secondly, I assume the procedure with calcium acetate can be done directly with phenyl acetic acid not its calcium salt, no?
That is not a good idea, as the phenylacetic acid may then react with the calcium acetate to form calcium phenylacetate and free acetic acid, which under the temperatures employed will evaporate and escape.
A lowered ratio between the Ca-Phenylacetate/Ca-Acetate leads to an increased formation of undesired dibenzyl ketone at the expense of less phenylacetone. The loss of acetic acid might be compensated for by the addition of more calcium acetate to the mixture, but I fail to see the advantage of using such a workaround compared to preparinging the proper calcium phenylacetate salt beforehand.
Also does anybody else think some of these are a bit impractical except as a possible laboratory curiosity?
Surely a lot of the procedures are obsolete and byzantine, but this compilation is intended to be comprehensive, rather than strictly practical.
* Some US states regulates 1-Phenyl-2-nitropropene though, see http://www.state.co.us/gov_dir/leg_dir/o
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
09-01-04 22:54
No 528927
OK Rhodium I surely would_ve said nothing when you just would've told me to not play the moderator, as I can understand this quite well; but since you tried to convey the impression that I said something wrong, I simply have to reply on that! Here we go...
About "Check your facts":
The "fact" I gave was: "Why do you think a pretty large number of bees use to prepare their own nitrostyrenes!? Better don't try to obtain them through ofiicial sources!"
The "fact" you gave is: "It is not explicitly regulated on a national level as far as I know*, but due to the noxious and unstable nature of nitroalkenes (not to mention a rather low licit demand), they are seldom offered for sale from chem suppliers. But even if the sale of 1-Phenyl-2-nitropropene isn't explicitly prohibited, it sure is a suspicious item and commercial aquisition should be avoided."
Now tell me the difference (besides redundant additional info about the noxious nature of nitropropenes and the low licit demand thing)!
(BTW sorry to interrupt Aurelius' compilation with that;
it doesn't seem to matter anyway)
And about "leave the moderation to the moderators":
WOW! whatta rating! Never seen that before... - but why do you think I would moderate something by stating "I think someone has gone off-topic"?
Greetz A
Pleased to meet you hope you get my name.
But whats puzzlin you is the nature of my game!
(Chief Bee)
09-02-04 01:58
No 528983
Now tell me the difference (besides redundant additional info about the noxious nature of nitropropenes and the low licit demand thing)!
The difference is that you did not answer the question directly - you replied with a question and gave some general advice. What you wrote might imply that 1-phenyl-2-nitropropene actually would be a regulated precursor, which it isn't in most parts of the world, including the EU and in US Federal legislation - just as I wrote in my post above.
My reply also included an example of an exception to this general rule, as well as offering an explanation as to why 1-phenyl-2-nitropropene seldom is found commercially, even though it is not explicitly prohibited to sell.
(BTW sorry to interrupt Aurelius' compilation with that; it doesn't seem to matter anyway)
If you have any further comments, please PM me - this is not the place for a discussion like this. If you are going to publicly argue about every rating you get in any of the forums, I will have to remove your posting privilegies in the chemistry forums, as you are severely cluttering them with posts which constantly are on the verge of being fully insignificant - your two latest posts in this thread does not really add much substance to the topic at all...
[Note to all: These off-topic posts will be removed in due time, as to not clutter this compilation forever...]
WOW! whatta rating! Never seen that before... - but why do you think I would moderate something by stating "I think someone has gone off-topic"?
I believe it is because you want to boost your ego by telling even fresher newbees than yourself about what you feel is appropriate behaviour around here. However, such advice is solely up to moderators and admins to dole out, as those are the only ones who are in a position to decide about appropriate bee-haviour.
If I catch you arguing about ratings or other severely off topic things in the chemistry forums again, prepare to have your posting privilegies suspended for a time. Ratings are off topic to discuss in all the chemistry forums, and should only be discussed in General/Couch/PM, if at all.
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
09-03-04 07:37
No 529281
A lowered ratio between the Ca-Phenylacetate/Ca-Acetate leads to an increased formation of undesired dibenzyl ketone at the expense of less phenylacetone.
Rhodium, do you think that an even greater ratio of Ca-Acetate: Ca-phenylacetate than was reported in the page at your site, in experiment 5, would increase the yield of P2P further?
Also, do you think replacing the CO2 stream with vacuum, to remove the ketones as they are formed would slow the reaction, or cause it to require higher temperatures?
The boiling sulfur bath means 444C, right?
(Chief Bee)
09-03-04 20:40
No 529391
Rhodium, do you think that an even greater ratio of Ca-Acetate: Ca-phenylacetate than was reported in the page at your site, in experiment 5, would increase the yield of P2P further?
I'd say that a further small increase might be possible, but not much. Compare with the table with the PAA:Ac2O ratios at ../rhodium /phenyla
Now the calcium salt pyrolysis is not exactly the same as the one refluxing PAA:Ac2O:NaOAc - but the general trend should still be the same. The highest ratio (w/w) between Ca-Acetate:Ca-phenylacetate used in the article is 1:1 - if you try the reaction with a 2:1 ratio you will see if that favors higher yield. At some point the small yield increase of P2P is not worth the added work (or cost) of a higher salt ratio, so don't overdo it.
Also, do you think replacing the CO2 stream with vacuum, to remove the ketones as they are formed would slow the reaction, or cause it to require higher temperatures?
The inert gas cannot be replaces with a vacuum, as the evaporation rate (or the internal temp) would be screwed up at lower pressures. Feel free to exchange the CO2 for any other inert gas though, such as He, Ar or N2.
The boiling sulfur bath means 444C, right?
Yes, I believe so.
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
09-05-04 14:22
No 529678
Thanks Rhodium.
That's unfortunate, the vacuum would evacuate it much faster out of the danger zone.
Rhodium this is from Xtaldocs interpretation of the Pb variation:
Preliminary experiments showed that the reaction itself initiated at a lower temp than that which was sustained during the roast. So, the first modification was to run it under a partial vacuum (5" Hg suction on a standard gauge).
So a very small vacuum might be ok? ![]()
Do you know how the barium reaction temperature is likely to compare with the calcium and Pb?
(Chief Bee)
09-05-04 19:32
No 529704
So a very small vacuum might be ok?
And a very small vacuum that is... Xtaldoc seems to refer to a relative pressure drop of only 5" from atmospherical pressure - a mere 633 mmHg (1 atm = 760 mmHg). I'd say that such a vacuum is fully OK to use at it will only facilitate the vapor flow, not change the bp noticeably.
Do you know how the barium reaction temperature is likely to compare with the calcium and Pb?
Seeing that the properties of Barium salts are far more similar to Calcium salts than to Lead(II) salts, I'd wager that the reaction temperature using Barium salts would be inbetween the two, but probably closer to 444°C than to the mere ~200°C required by the Lead(II) method. As Barium is a lot more toxic than calcium, there is no real advantage to the use of such salts.
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
09-07-04 13:16
No 530107
And a very small vacuum that is... Xtaldoc seems to refer to a relative pressure drop of only 5" from atmospherical pressure - a mere 633 mmHg (1 atm = 760 mmHg). I'd say that such a vacuum is fully OK to use at it will only facilitate the vapor flow, not change the bp noticeably.
It is small, and of course there's no guarantee it will work the same for the calcium variation.
A combination of inert gas and vacuum might also be a possible option. or just the inert gas like the write-up says.(going around in circles)
Seeing that the properties of Barium salts are far more similar to Calcium salts than to Lead(II) salts, I'd wager that the reaction temperature using Barium salts would be inbetween the two, but probably closer to 444°C than to the mere ~200°C required by the Lead(II) method. As Barium is a lot more toxic than calcium, there is no real advantage to the use of such salts.
That makes sense. Plus, CaCO3 is a lot cheaper.
(Chief Bee)
10-04-04 00:31
No 534323
(Rated as: excellent)
Here follows several japanese syntheses of phenylacetone, as summarized in Chemical Abstracts.
CA Nomenclature note: Analogous to the shorthand Ac for Acetyl (Me[/sub]CO-), Bz is short for Benzoyl (PhCO-) and not Benzyl (PhCH2-)
Reaction of α-halocarbonyl compounds with Grignard reagents. II. Reactions of α-chloroacetophenones.
Teiichi Ando, CA 54. 449215 [Yuki Gosei Kagaku Kyokaishi 17, 777-82 (1959)]
Reactions of some α-chloroacetophenones (RCOCH2Cl) with Grignard reagents in ratio of 1:1 and 1:2 were studied and compared (cf. CA 53, 17971d)
Ratio of 1:1: To a Grignard reagent prepd. from 17.3g PhBr, 2.7 g Mg, and 100 mL Et2O was dropped 15.6g α-chloroacetophenone (I) in 100 mL C6H6 at -3°C to 0°C, the mixt. stirred at 0°C for 30 min, concd. till the bp of the mixt. increased to 72°C, and then refluxed 4 hrs., cooled, decompd. with ice and HCl, and the sepd. org. solvent layer distd. to give 40% deoxybenzoin, bp 160-170°C/6 mmHg, mp 55-56°C (MeOH); semicarbazone mp 147-148°C. Similarly were carried out the following reactions. I with MeMgI, 45% phenylacetone (101-104°C/23mmHg; semicarbazone m. 189-190°C). α-chloro-p-fluoroacetophenone (III) with MeMgI, 41% p-fluorophenylacetone (120-130°C/30mmHg; semicarbazone mp 199-200°C).
Ratio of 1:2: I (7.7g) in 100 mL C6H6 was treated with Grignard reagent prepd. from 17.3g PhBr, 2.7g Mg, and 100 mL Et2O to give 44% 1,2,2-triphenylethanol, mp 87-94°C (ligroin), and 1.8g triphenylethylene, bp10 200-120°C, mp 67-68°C (EtOH). Similarly were carried out the reaction between III with p-F-PhMgBr. 72% tris(p-fluorophenyl)ethylene (bp 195-205°C/12 mmHg, mp 71-72°C).
These results showed that ketones were obtained when the molar ratio of the reactants was 1:1, and 1,1,2-trisubstituted ethanols were obtained when that ratio was 1:2. Based on these experimental results and on the conception of migratory aptitude of atomic groups, the mechanism of the reaction was also discussed.
______ _____ ____ ___ __ _
Reduction of 1-phenyl-1,2-propanedione. I. Synthesis of phenylacetone.
Genshun Sunagawa and Kichihei Okuda, CA 46, 11146a(1952) [J. Pharm. Soc. Japan 72, 117-118 (1952)]
Catalytic reduction of 18.5 g BzAc (I) in 40 mL MeOH with PtO2 gives 11.9 g. PhCH(OH)CH(OH)Me (II) mp 92-93°C; catalytic reduction of a mixt. of I and MeNH2 in MeOH with PtO2 gives dl-ephedrine·HCl, and the mother liquor taken up with ether and distd. gives II.
Refluxing 20 g II with 200 g 20% H2SO4 5-7 h, taking up with ether, and distg. give 13g PhCH2Ac; semicarbazone, mp 190-6°C.
______ _____ ____ ___ __ _
Phenylethynylcarbinol and its derivatives. II. Synthesis of phenylacetone
Ichiro Hirao, Chem. Abs. 10534c (1954) [J. Chem Soc. Japan, Ind. Chem. Sect. 56, 265-266(1953)]
Commercial PhCH(OH)CCH was hydrated with H2SO4 to PhCH(OH)COCH3, which was hydrogenated with Raney-Ni catalyst by H2 of 50 atms. in an autoclave at room temp. The white crystals obtained correspond to 1,2-dihydroxy-1-phenylpropane (I) of mp 91-92°C; yield, 95% PhCH2Ac was prepd. by heating of I with H2SO4, oxalic acid, H3PO4, etc.; yield approx. 73%.
______ _____ ____ ___ __ _
Synthesis of phenylacetone
Keiichi Shishido and Shigeru Kukita
CA 42, 6339g [J. Soc. Chem. Ind. Japan 48, 34(1945)]
A Grignard reagent prepd. from 6g Mg, 1.70 mL anhyd. ether, a small crystal of I2 and 32 g PhCH2Cl was dropped slowly into 20g AcCl in 150 mL ether, kept at -10°C to -15°C, and the product treated with ice-cooled concd. HCl, extd. with ether, neutralized, and distd. under 9 mmHg at 87-93°C, giving 27% PhCH2Ac, mp 188.5-189°C from alcohol).
This one is a little fishy - the mp data makes no sense, as P2P is a liquid. It does however jive with the mp of P2P semicarbazone, which is mentioned in two of the above abstracts as well. I wonder if it's a typo or a mistaken product?
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
10-05-04 00:59
No 534445
Am I reading this correctly that the Dione gives both ephedrine and P2P (via the Diol)? What would be the easiest way to make the Diol (1 Phenyl 1,2 Propanediol)?
.........Reduction of 1-phenyl-1,2-propanedione. I. Synthesis of phenylacetone.
Genshun Sunagawa and Kichihei Okuda, CA 46, 11146a(1952) [J. Pharm. Soc. Japan 72, 117-118 (1952)]
Catalytic reduction of 18.5 g BzAc (I) in 40 mL MeOH with PtO2 gives 11.9 g. PhCH(OH)CH(OH)Me (II) mp 92-93°C; catalytic reduction of a mixt. of I and MeNH2 in MeOH with PtO2 gives dl-ephedrine·HCl, and************************the mother liquor taken up with ether and distd. gives II.*****
Refluxing 20 g II with 200 g 20% H2SO4 5-7 h, taking up with ether, and distg. give 13g PhCH2Ac; semicarbazone, mp 190-6°C.
(«
 »)
»)10-05-04 04:02
No 534475
Take from the following thread:
Post 108587 (dormouse: "Two new MD-P2P synths for y'all to chew on! -psychokitty", Novel Discourse)
Author Topic: Two new MD-P2P synths for y'all to chew on!
psychokitty
Member posted 12-14-98 09:27 PM
----------------------------------------
The following information was taken from these three references:
J.C.S. Perkin I, 1974 pp1727-1731.
J.C.S. Perkin I, 1975 pp1548-1551.
Chemical Communications, 1971, pp818-819.
These experiments originally used methyleugenol to synthesize 3,4-DM-P2P, but so what? They should work equally well on safrole to make MD-P2P.
(a). Hydroxlation of methyeugenol to DM-P2Pol.
Methyleugenol (133mg) dissolved in tetrahydrofuran (3ml) was added to a solution of mercury(II)acetate (400mg) in tetrahydrofuran-water (1:2,4.5ml) and the mixture was stirred at room temperature for 24hr. After addition of 3M-sodium hydroxide (4ml) followed by 0.5M-sodium borohydride in 3M-sodium hydroxide (4ml), mercury was allowed to settle and filtered off; solid sodium chloride was then added to saturate the water layer. The upper layer and ethereal extracts (3 times 10ml) of the lower one were pooled, dried (Na2SO4), and evaporated. The residue (123mg), homogenous by GLC, was crystallized from ether-light petroleum (bp40-60degC)and shown to be DM-P2Pol.
(b) DM-P2Pol oxidation
DM-P2Pol was dissolved in acetone (1ml) and oxidized with an excess of Jones reagent at 0degC for 5min. After the addition of isopropyl alcohol (0.5ml) followed by saturated aqueous sodium hydrogen carbonate (2ml), the mixture was extracted with chloroform (5 times 10ml). The residue after evap. of solv. was DM-P2P (25mg).
The above synthesis is pretty cool, but in my opinion, inferior to the Wacker analogue reaction using mercuric acetate and Jones reagent that used to be #2 in TSI.
Now for the real goods:
Sythesis of DM-P2P directly from methyleugenol using PdCl2-LiCl/Hg(OAc)2/CuCl2.
Mercury(II)acetate (8.96g)and methyleugenol (5g) were stirred at 25degC for 20min in MeOH (180ml)and then added to a stirred solution of lithium chloride (238mg), palladium chloride (493mg), and copper(II)chloride (14.4g)in MeOH. The mixture was heated under reflux for 1hr, then poured into saturated sodium hydrogen carbonate solution (500ml); the solution was filtered and heated under reduced pressure to remove MeOH. The residual solution was extracted with ether (6 tms 100ml) and the extract was dried and evaporated. Distillation of the oil so obtained (5.2g) under vacuum afforded DM-P2P (3.6g), bp 120-123 at 0.6 mmHg.
Hey, Strike! I wanna be in the book!
The last synthesis above is pretty cool, no? MD-P2P in one hour from safrole. Only thing that suck is the amount of solvent needed for scaling up. Maybe it can be reduced. Anyone have any comments out there in da Hive?
Before I forget to mention, what is actually happening here is that the MeOH-HgOAc add to the double bond of the alkene and then the PdCl2 swaps places with the HgOAc, which forms an unstable intermediate that rearranges to the final ketone. I wonder what would happen if you used PdCl2 in the aminomercuration synth. Would the PdCl2 swap with the HgNO3, forming an unstable intermediate, to then eventually form -- what? MDMA?
Oh, well. It's just an idea.
(«
»)10-06-04 04:44
No 534615
As originally reported by Rhodium in the following thread:
Post 286777 (Rhodium: "p-Fluorophenyl-2-propanone", Novel Discourse)
and, in more general terms, by Regis in the following thread:
Post 365747 (Regis: "Arylmagnesium bromides/chloroketones reaction", Novel Discourse)
Some Fluorinated Amines of the Pressor Type
C. M. SUTER AND ARTHUR W. WESTON
JACS Vol. 63, pp. 602-605 (1941)
Experimental example of the Grignard synthesis of p-fluorobenzyl methyl ketone:
p-Fluorobenzyl Methyl Ketone.-A Grignard reagent was prepared from 35 g. (0.20 mole) of p-fluorobromobenzene and 4.6 g. (0.19 mole) of magnesium. To this was added 18.5 g. (0.2 mole) of chloroacetone in 50 ml. of ether as rapidly as the refluxing of the reaction mixture would allow. The ether was removed by heating the reaction flask in an oil-bath and at 100 'C the residue foamed with formation of a gel from which ether was removed slowly by heating at 135-140'C for forty-five minutes. The flask was cooled, ice and dilute acid were added, the heavy oil which separated was removed with ether and the ether solution dried and fractionated. There was obtained 11.2 g. (37%) of practically pure ketone distilling at 106-107'C (18 mm.).
A variation of this procedure in which the reaction mixture was not heated above 100'C gave a product that could not be satisfactorily fractionated. Substantially pure ketone was finally obtained by conversion to the sodium bisulfite addition compound followed by regeneration with dilute sodium carbonate. It distilled at 108'C (18 mm.) as a light yellow oil with +'D 1.4965, d*O, 1.107, MD calcd. 40.07, obsd. 40.17. After a few days of standing, crystals of p-fluorobenzoic acid were deposited. Carbon and hydrogen analyses gave low results even with material kept in a sealed ampoule. A solid derivative was therefore prepared. The dinitrophenylhydrazone did not form but the semicarbazone, m. p. 200.5-201.5', separated readily. Analysis by the Jamieson method of titrating with potassium iodate gave only fair results. Anal. Calcd. for C10H12ON3F: eq. wt., 52.30. Found: eq. wt. 51.50. In this analysis the end-point was not stable, probably due to the reaction of the liberated ketone with iodine.
(Moderator)
10-16-04 02:52
No 536024
(Rated as: good read)
Another old p2p synthesis, a Claisen condensation of phenylacetyl chloride using zinc-methyl iodide in toluene-ethyl acetate yielding 72%; Ber 58 339-41 (1925)

Chemistry is our Covalent Bond
(Wonderful Personality)
10-16-04 10:10
No 536062
Translation:
Preparation of methylbenzylketone:
54g phenylacetylchloride dissolved in the same volume of toluene was added to a solution of methyl-zink-iodide, which was prepared from 32,5g zinc (as zinc/copper couple) and 71g methyliodide in toluene and 16ccm ethylacetate ("Essigester"). Yield: 34g, 72% of theory.
Earlier in the article the preparation of phenylacetylchloride is mentioned:
...was prepared in quantitative yields from phenylacetic acid and thionylchloride....
P2P is only a precursor in the experiments of the authors and so they keep it short.
Earlier it is mentioned that hydrolysis of 3-phenyl-propionylacetone, C2H5.CO.CH(C6H5).CO.CH3 with 20% aqueous NaOH solution yields a mixture of ethyl- and methyl- benzylketone
so near, so far......