(Hive Bee)
01-19-04 05:24
No 483260
(Rated as: excellent)
SWIM finally tried 4-fluoroamphetamine, and was very impressed with the stuff! Here's how it happened:
All chemicals used were reagent grade except for zinc, which was technical grade. This was a proof-of-method run, and as you'll see there were a couple of fuckups along the way, though ones which can be easily avoided from now on. There are pictures from various points along the way, but SWIM doesn't know how to get them up for viewing in here. Anybody?
1-(4-Fluorophenyl)-2-nitropropene:
6.05 mL of 4-fluorobenzaldehyde (56.4 mmol) was added to a 100 mL round bottomed flask containing 4.26 mL of nitroethane (59.2 mmol). 0.73 mL of 40% methylamine (8.46 mmol) was added to the stirring mixture, and the flask was heated at ~60°C. After about an hour, the flask was cooled to room temperature and 0.81 mL of glacial acetic acid (14.1 mmol) with 50 mL deionized water as added. The flask was then chilled in a freezer, but the light-yellow solution did not crystallize. TLC showed a decent amount of 4-fluorobenzaldehyde remaining, so additional water was added, the flask was swirled vigorously, and once the layers separated the light yellow organic layer was isolated and placed in a round bottom flask fitted with a reflux condensor. Another 4.26 mL of nitroethane (8.46 mmol) was added, followed by 0.25 mL of n-butylamine. The solution was held at reflux for 3 hours forming a dark reddish solution, which was cooled to room temperature and then placed in the freezer. After an hour the mixture was filled with yellow-orange solid. After filtering and recrystalizing the solid from boiling isopropyl alcohol, there was 5.63 g of 4-fluorophenyl-2-nitropropene (31.1 mmol, 55% yield) as shiny yellow spikes, some of which were over an inch long.
1-(4-Flurophenyl)-2-nitropropane:
Within a day the tips of the crystals had turned slightly orange indicating some decomposition, so it's probably best to use it immediately. 2.35 g of old, slightly-chunky sodium borohydride (62.2 mmol) was dissolved in vigorously stirring 30 mL isopropyl alcohol and 10 mL water. The 5.63 g of nitropropene was added in small increments, causing strong foaming and heat generation with each addition, followed by complete loss of yellow color shortly thereafter. Unfortunately, during the addition one portion was added a little too quickly and a small foam-volcano erupted out of the flask...... causing some loss. After the rest of the borohydride had been added (whole addition took about 1 hour), it was left to stir for 30 minutes. Still there remained some yellow color. Additional isopropyl alcohol/water was added, and more borohydride was added by the spatula-full until most of the yellow color had disappeared. At least an additional 2 g of borohydride was added, but actual amount not followed. It is likely that the borohydride used was of degraded quality. After an additional hour of stirring, all yellow color was gone. The slurry was filtered through celite (which was washed with additional isopropyl alcohol/water) giving a colorless solution. Solid sodium chloride was added until the solution was saturated and an alcohol layer formed. The alcohol layer was separated, and showed one spot by TLC.
1-(4-Fluorophenyl)-2-aminopropane Hydrochloride:
The Zn/HCOOH reduction may have been done in excess... Comments? The alcohol layer was placed in a round bottom flask and 21.1 g of zinc dust (tech grade, 322 mmol) was added. The solution was stirred vigorously and a reflux condensor was attached. 36.7 mL 90% HCOOH (966 mmol) was slowly added down the condensor over about an hour, causing mild foaming and excessive heat generation; no external heat control was used. Stirring was continued until the mixture returned to room temperature. The zinc was filtered through celite, giving a pink (or perhaps really light red) solution. The filtrate was reduced to about 30 mL by boiling away the isopropyl alcohol at atmospheric pressure. 10% HCl was then added until pH was 1-2. The solution was poured into a separatory funnel and washed with 3 x 25 mL toluene, which removed all of the pink color. The aqueous layer was then basified to pH 12 using high-concentration NaOH solution, occaisionally adding additional water to try to keep everything dissolved. A yellow oil floated to the top of the solution, which was poured into a separatory funnel containing 50 mL toluene. The two-phase mixture was left in the dark for about 2 days before it was separated. The aqueous phase was extracted 2 additional times with 50 mL toluene. The combined toluene extracts were washed with 2 x 25 mL water and an additional 25 mL of brine. The 4-fluoroamphetamine was extracted with aqueous HCl (25 mL water, conc. HCl was added dropwise until shaking gave an acidic aqueous layer). The water was evaporated to give a chunky slightly-yellow solid. The solid was dried under heat lamp, crushed to a very fine powder, and rinsed with ice-cold acetone repeatedly until all yellow had disappeared from the solid. Drying under a heat lamp gave 2.78 g 4-fluoroamphetamine HCl (14.7 mmol, 47% yield) as a powdery white solid.
Bioassay:
This stuff outdid SWIM's expectations. Very nice! 140mg material was eaten, and took about 45 minutes to onset. There's been some reports that say it is "somewhere between MDMA and meth" which is a pretty dead-on statement. A bit of background, SWIM is a recovering meth addict and currently taking zoloft. SWIM also "lost the magic" some time ago with MDMA, and wasn't really expecting a whole lot from the 4-fluoroamphetamine. Once it kicked in however, there was a very strong "nice-head" feeling, pretty euphoric like MDMA. Also, a distinct stimulation, though this stuff felt rather less harsh on the body than standard amphetamine sulfate. It had an excellent energetic body high similar to meth, and promoted lots of talking. The sexual drive wasn't as strong as with meth however. The nice head-high and slight lowering of inhibitions were very MDMA-like, and pretty fun. Strong worthwhile head- and body high went for a good 4-5 hours, afer which it slowly passed leaving a more or less standard amphetamine high for several more hours. After about 10 hours, some valium was taken and sleep wasn't too hard to accomplish. Felt just tired the next day, not too braindead. The day after was IMO not as bad as that of MDMA's day after.
From a few other testings it has been determined that a boosting dose (of say 50 mg) about 2 hours into the high is alright, but trying to redose at any amount once you begin to come down just produces prolonged and less comfortable stimulation lacking the real fun of the drug.
SWIM was quite pleased at the yield, considering the small volcano that happened accidentally during reduction. This synthesis is a pretty convenient one, not hard to carry out at any point.
There ya go.
-SpicyBrown
(Chief Bee)
01-19-04 09:04
No 483287
Excellent report! Any pictures you can email to me and I'll put them on my site.
The Hive - Clandestine Chemists Without Borders
(Active Asperger Archivist)
01-19-04 19:24
No 483341
Post 353421 (malvaxman: "4-bromoamphetamine", Chemistry Discourse)
States that p-haloamphetamines are have been shown to cause neurotoxicity (probably death of cells). F-amphetamine was specifically mentioned as NOT being used for this purpose, but did you (Barium) mean that F-amphetamine is safe? Rhodium, you mention Br, Cl, and I-amphetamines as nasties, but don't specifically mention how safe F-amphetamine is or might be.
Could somebody clear this up? (I suppose this is excessive as the clues are pretty straight forward, but I'd like to retain my brain cells in whatever fashion my hobbies will allow.)
Act quickly or not at all.
(Hive Bee)
01-19-04 20:52
No 483355
Just realized SWIM forgot to mention one minor step: following removal of the solid zinc by filtration, the volume of the filtrate was reduced substantially by boiling at atmospheric pressure. The original post has been edited to include this.
Rhodium: Thanks! Pictures will be off to you shortly.
-SpicyBrown
(Chief Bee)
01-19-04 21:02
No 483358
I state it in my 4-Fluoroamphetamine Doc (../rhodium /fluoro
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
02-05-04 15:01
No 486574
(Rated as: good read)
The initial condensation attempt with methylamine, which wasn't very successful:
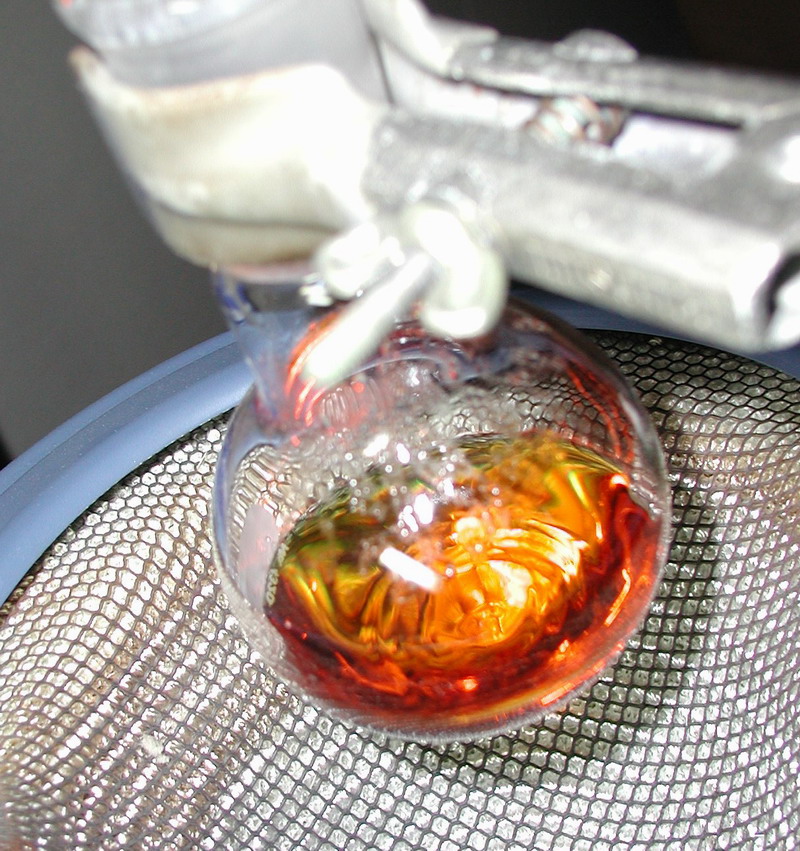
The condensation after washing previous reaction mixture and using n-butylamine instead:
Crude nitropropene crystallized spontaneously:

The recrystalized nitropropene looked quite nice:

Freebase 4-fluoroamphetamine floats out of basic solution:

And finally, ~2.8 g 4-fluoroamphetamine HCl:

-SpicyBrown
(Stranger)
02-08-04 03:44
No 487183
Spicy, if you dont mind me asking, how much would it cost if one were to replicate the same procedure, using the same quantities?
Just trying to figure out how much 2.8g of end product would cost to make! Swim has the oppurtunity to buy it, so i want to know how much markup the bastards are applying!
The devil made me do it.
(Hive Bee)
02-08-04 07:17
No 487246
Spicy, if you dont mind me asking, how much would it cost if one were to replicate the same procedure, using the same quantities?
You know, I'm honestly not sure. A lot of the chemicals involved weren't purchased specifically for this project- For example, I bought 200-some odd grams of NaBH4 a long time ago, which wasn't cheap, but this synthesis used only a small portion of that. Solvents and related chemicals were also already on hand. Other reagents such as the 4-fluorobenzaldehyde itself and nitroethane, were basically acquired through trade with people that have better connections then I.
I do know one thing though, the cost of producing a mere 2.8 g would be comparatively outrageous compared to the price of producing say, 500 g, which is probably how wherever you may buy it from is making it.
Sorry!
-SpicyBrown
(Old P2P Cook)
02-08-04 08:54
No 487258
What does the freebase smell like. Any different from methamphetamine or MDMA freebase?
All those moments will be lost in time, like tears in rain.
(Hive Bee)
02-08-04 21:27
No 487330
Terbium, I'd say it smelled a good deal like methamphetamine freebase. I haven't actually smelled MDMA freebase (unfortunately), so I can't make that comparison.
-SpicyBrown
(Stranger)
02-10-04 00:07
No 487546
is either nothing or safrol/piperonal like...A little bit sweeter when compared to P2P.
I´m not suffering from Magnon´s Syndrome...It just itches!
(The Archetypical "Good Guy")
02-20-04 13:01
No 489952
(Rated as: good read)
Because of the low yields reported by people on the condensation of nitroethane and p-F-Benzaldehyde, I decided to try a few tweaks to boost the yields. Unfortunately it did not help
General theory:
p-F-benzaldehyde is condensed with nitroethane in the standard Henry reaction. SpicyBrown and others who has tried this using butylamine as the catalyst never reported more than 55% yield, which is somewhat on the low side, considering the price of this particular benzaldehyde. Aqueous methylamine seemed to perform even worse. In the henry reaction one mole of water is formed per mole of R-nitropropene formed. In order to push the equilibrium as far as possible to the right, i decided to test how anhydrous conditions would perform by removing the water during the reaction as it is formed, using molecular sieves. As catalyst a 2M ethylamine in THF solution was used (no particular reason for this - it was simply available at the time). The whole reaction is run in IPA (3A sieves should be compatible with everything here, AFAIK...) One mole of water is formed per mole of benzaldehyde used (assuming quantitative yields of course). One mole of water equals 18 grammes. 3A molecular sieves are able to bind 21% wt/wt of water per mass of sieves used. Thus 86 g's of 3A mol. sieves are required per mole of substrate. Assuming that a little water is present in the reactants 100 g's is probably better to use.
Experimental:
Reagents used:
12,4 g (100 mmole, MW=124 g/ mole) p-fluoro-benzaldehyde
8,25 g (110 mmole, MW=75 g/mole) nitroethane
10 mL (20 mmole, MW=45 g/mole) 2M EtNH2 in THF
25 mL 99% IPA
10.5 g 3A mol. sieves
The Reaction:
The benzaldehyde was mixed with the nitroethane in a erlenmeyer flask, whereafter both of the measuring beakers where rinsed with 12.5 mL's IPA and poured into the reaction flask. 10 mL 2M EtNH2 in dry THF (smelly crap) was added using a glass pipette. Finally the molecular sieves where added in one portion. The solution was totally transparent and colourloess at this point. A magnet was dumped into the mixture, and the whole shebang was lowered into an oil bath at 60 degrees and stirred gently, to avoid destroying the sieves.
At t+10 mins: the reaction mixture had taken on a canary yellow look but still reaked of benzaldehyde.
At t+30 mins: the colour of the mixture was somewhat darker and the benzaldehyde smell was only faint.
At t+60 mins: the colour did not go any darker and the benzaldehyde smell was nearly gone.
At t+120 mins: No change from t+60
At t+180 mins: No change from t+60, 1,35 g's 85% formic acid added to neutralize the catalyst. Flask removed from oil bath.
At t+181 mins: Mixture filtered.
The flask was removed at t+180 mins, and the post reaction mix was suction filtered while still hot. The filtered sieves where rinsed with a minimal amount of IPA. The sieves where put in a brown jar for future regeneration.
The flask containing the product was placed in the freezer over night. In the morning the bottom of the flask was covered in pretty canary yellow needle like crystals. A few of the crystals where removed from the flask and saved in the freezer in case a later crystallization would prove difficult. Most of the solvent where stripped from the post reaction mixture on a waterbath under aspirator vacuum. A solid cake remained after refridgiating. This was recrystallized in boiling IPA and filtered yet again.
The yield was a mere 6.61 g of p-fluoro-nitropropene, which corresponds to a 38% molar yield
Conclusion:
Seems that this particular molecule wants a long alkyl chain on the catalyst during the condensation. Even if the anhydrous conditions did help, it sure wasn't alot. Oh we'll it's not a complete waste as there has been previous discussions here on the-hive if more dry conditions for the Henry condensation will improve the yields. We'll this experiment has shown that if they do so, it is very miniscule. I will repeat the experiment once i get my hands on some n-butylamine, such that i have a control reference in SpicyBrowns trials.
The nitropropene was reduced in the usual manner to the nitropropane. The IPA layer from the reduction will be used directly in a Barium Pd/C - KCOOH CTH reduction to see if this will give better yields than what SpicyBrown reported with Zn/HCOOH.
Regards
Bandil
Nuke the whales!
(Hive Bee)
02-21-04 01:49
No 490067
Bandil, excellent! Even if the results weren't exactly happy, I know for sure now that for my next attempt I'll stick with the n-butylamine; I had been considering trying some other things. I may still however try a long-term room-temp condensation with n-butylamine, as such conditions for ~20 days seem to give exceptional yields with piperonal; it may be worth the wait for this substrate if the yields are equally good.
I'm eager to hear how the Pd/C-KCOOH reduction goes- if it seems to work well that could be one use for my oh-so-precious Pd/C I've been saving for a special occaision...
-SpicyBrown
(The Archetypical "Good Guy")
02-21-04 20:14
No 490185
The CTH reduction ran super smooth and without a glitch
Oh my god CTH reductions are nice to work with. This has been one of those "aha moments" we all treasure so much
Nuke the whales!
(Hive Bee)
02-22-04 17:18
No 490389
Very nice work ;)
Bandil, if you find some time a writeup on the CTH-Reduction would be great!
best wishes,
xicori
(The Archetypical "Good Guy")
02-23-04 09:44
No 490561
The weekend got a little crazy, so i didn't got any work done
I'll work everything up tonight, and post the writeup about the NaBH4 / CTH reduction tomorrow!
Regards
Bandil
Nuke the whales!
(The Archetypical "Good Guy")
03-05-04 13:16
No 493132
The reduction using CTH was just dandy
Unfortunately the HCl of this amphetamine seems very hard to form... It goes into some strange liquid-salty form when recrystallization is attempted. Thus the yield is very hard to determine.
I'll try it again with 4-fluoroamphetamine and 2-fluoroamphetamine and form the sulfates following!
Regards
Bandil
Nuke the whales!
(Chief Bee)
03-05-04 14:39
No 493148
I have now posted your writeups on my page:
../rhodium /pfa.spi
../rhodium /4f-p2np
And also rewritten the chemistry section of ../rhodium /fluoroa
The Hive - Clandestine Chemists Without Borders
(Newbee)
07-09-04 04:44
No 518313
I swim want to dream about this using only methylamine as the catalyst for the first reaction, avoiding n-butylamine...
1- Which quantity of methylamine would be needed ?
2- What about expected yield ?
(Hive Adickt)
07-09-04 07:05
No 518333
UTFSE!!!!
Or read this one
Post 422844 (Bandil: "Verification of Bariums research", Methods Discourse)
(Newbee)
07-09-04 08:14
No 518337
Well
According to Post 422844 (Bandil: "Verification of Bariums research", Methods Discourse), everything should be fine. Ratios are all in line with what Bandil wrote.
BUT:
the light-yellow solution did not crystallize ... so additional water was added ... Another 4.26 mL of nitroethane (8.46 mmol) was added, followed by 0.25 mL of n-butylamine
Meaning that for this very specific aldehyde the catalyst (methylamine) FAILED to complete the reaction...
WHY ????
(The Archetypical "Good Guy")
07-09-04 09:39
No 518349
grandpa>
In my post Post 422844 (Bandil: "Verification of Bariums research", Methods Discourse), 2,5-dimethoxybenzaldehyde was used as the substrate. This is highly activate compared to 4-fluorobenzaldehyde, and gives much higher yields in the condensation reactions. That is most likely the reason that methylamine failed with 4-fluorobenzaldehyde.
Generally speaking its a good idea to use a N-alkylamine (-propyl or -butyl) in a small excess of GAA (compared to the catalst) for the condensation, if you are working with the not so activated substrates.
Kind regards
Bandil
Nuke the whales!