12-13-00 21:45
No 75319
Has anyone had any success W/ the
bromosafrole=safrole-azide=mda route
described in TS2? the only post i have
seen is the one submitted by ritter
MDA via 1 pot 1.1,Is there any other posts concerning
Sodium Azide
(Chief Bee)
12-14-00 03:20
No 75342
Trust Ritter. He is a very experienced chemist.
http://rhodium.lycaeum.org
(Stranger)
12-14-00 10:14
No 75392
I wanna trust ritter,but the only problem i have
is w/ the size of the reaction.In strike's reaction he uses
120g bromosafrole,compared too 24g in ritter's reaction.
it seems to me Strike's recipe is a little simpler.
i have 2 ??? to ask:
1st:the msds for sodium azide says it is incompatible
w/ bromine & w/ h20,distilled of course
2nd: both of these are called for in the 1st
stage of the reaction converting bromosafrole
into safrole azide,are we gonna have an
explosion here or should i just trust the
science?
"RHodium,Ritter,STRIKE"
Talk to me johnny!!!!!!
(Chief Bee)
12-14-00 11:53
No 75410
It doesn't matter if sodium azide is incompatible with bromine, because there is no bromine in the reaction mixture. The bromosafrole will liberate bromide ion, but that is something completely different. There is no problem mixing NaN3 with water either, the reason the MSDS says otherwise may be because prolonged storage of NaN3 in water may cause partial (non-violent) breakdown of it.
The reaction used in TSII is indeed simpler, but gives lower yields, and requires vacuum distilllation of the safrole azide, which is potentially dangerous due to the inherent instability of organic azides.
http://rhodium.lycaeum.org
(Wizard Master)
12-23-00 17:52
No 77514
Add bromosafrole (scarlet wine red colour) to a solution saturated with sodium azide in 95% ethanol or 50/50% ethanol-water. Warm to 50 deg C with magnetic stirring and continue until all the bromosafrole (scarlet wine red colour) has turned a golden amber colour (1-2 hours)[1].
Reduce with sodium borohydride.
[1] OR separate the MDP-2-azidoP and then reduce with sodium borohydride.
(Hive Bee)
01-02-01 10:26
No 79337
Happy New Year Bees!!
improv_chem managed to think a little between brain cell assults this vacation.
Could you seperate the MDP-2-azidoP and reduce it in an electric cell??
SWIM is still looking for some NaBH4....
Azide -> amine?? seems like a simple thing to do, could a cell do it?
It's all just a dream, I hope i don't wake up because of that incessent buzzing sound...
(Wizard Master)
01-02-01 18:20
No 79426
Use Al-Hg reduction in isopropanol + 5% water, or 95% ethanol.
(Hive Bee)
01-04-01 06:28
No 79815
WizardX-> sounds real good has anyone actually tried this??
Shit, guess SWIM's gonna have to make some more HBr to play with. Will HBr dissolve teflon plumbing tape?
Got a little leak in SWIM's acid distillation apparatus ...
SWIM is real happy he obtained that NaN3 a while back. Had no idea it could be that useful a chemical.
If SWIM gets this to work he will let you all know. (might be a while, SWIM's sass supplier is an idiot!)
thanks for the tips;
--improv
It's all just a dream, I hope i don't wake up because of that incessent buzzing sound...
(Hive Bee)
01-06-01 12:36
No 80366
Well, i answered my own question: HBr does not dissolve teflon tape. After a nasty 2 hour session SWIM has a bunch of 48% HBr from some pool sanitizer(NaBr) and some drain cleaner.
Here's some stuff from the old board that might be useful to someone:
Safrole-azide:
"In a one-neck flat-bottom flask on a stirplate[turned on] are combined 100g bromosafrole , 30ml dH2O and 50g NaN3. 5g butylamine are dripped in. Then a condenser is attached, the soln brought to reflux and kept there for 6h. After cooling, extraxt with Et2O, dry thru Na2SO4 and distill off the ether to get the Safrole-azide. Yield is 75%. You can get near 100% if you take 'aliquat 336' as PTC. without a PTC yield is only 50%.
Reduction:
200g safrole-azide is dissolved in 1000ml MeOH and 100g SnCl2 [or the dihydrate] are scraped in in small increments.Almost imediately heavy bubbling and heat will evolve. This is released N2, tells you that everything works. Then let stir for another 60mins. Remove the MeOH by vacuum distillation. To the remaining oil add 500ml ice cold dH2O. Bring the pH to 9-10 by addition of dilute NaOH soln. Saturate with NaCl. Extract with ether, wash ether extract with NaCl soln and dry thru Na2SO4. Remove solvent by distillation. Yield is 98%."
I like that SnCl2 reduction method, looks real simple if you have some SnCl2.
WizardX: your recipe uses EtOH as the solvent for making the safrole azide at 50C. Could this rxn be done at 65C in refluxing MeOH? Should not make a difference if it is MeOH or EtOH, or even IPA right?
Any specific reason for using the ethanol?
Would the bromosafrole still dissolve if 50% EtOH was used or would it be a 2-phase thing?
Shit it's getting kinda late and I need a coffee.
Any tips on the solvent issue appriciated;
It's all just a dream, I hope i don't wake up because of that incessent buzzing sound...
(Wizard Master)
01-21-01 18:49
No 168758
I like that SnCl2 reduction method, looks real simple if you have some SnCl2. Becareful not to form Sn-NH2 complexes (lignates)
WizardX: your recipe uses EtOH as the solvent for making the safrole azide at 50C. Could this rxn be done at 65C in refluxing MeOH? Should not make a difference if it is MeOH or EtOH, or even IPA right? YES it's OK. Make no real difference
Any specific reason for using the ethanol? NO reason, cheap
Would the bromosafrole still dissolve if 50% EtOH was used or would it be a 2-phase thing? Dissolve
(Stranger)
06-22-02 18:12
No 324231
I found this article today. Is it usefull?
---------------------------------
A Practical Procedure for the Synthesis of Alkyl Azides at Ambient Temperature in Dimethyl Sulfoxide in High Purity and Yield
Salvador G. Alvarez*, Miho T. Alvarez
*Department of Chemistry and Biochemistry, University of California, Santa Cruz, CA 95064, USA, Fax +1(408)4592935
Described is the often neglected, but very efficient, procedure for the preparation of primary and secondary alkyl and cycloalkyl azides in excellent purity and high yields. At ambient temperature conditions, the alkyl azides were readily obtained from alkyl bromides by the nucleophilic substitution of bromide with 1.2 equivalents of sodium azide in dimethyl sulfoxide (DMSO). This facile and practical procedure provides highly pure products and eliminates the hazards associated with distillation of alkyl azides.
Alkyl azides are prepared from alkyl bromides and NaN3/DMSO
----------------------------------
you can get the full article + procedure w yeild table from here:
http://www.thieme-connect.com
(search journals for "alkyl azides")
gotta sign up for a free month or whatever takes 2 seconds no CC.
Any reason why SW shouldn't try this with his bromosafrole?
(Master Whacker)
06-23-02 17:26
No 324511
One little piece of info I can relate concerning the use of DMSO as solvent is that if you decide to run the rxn at high temp, nothing but poisonous garbage will result. It's nice to know that the secret is to run the rxn. at room temp. Thanks for digging that up endo1 !
(Stranger)
06-23-02 18:11
No 324541
Thanks for the info Ritter. It was your original method on Rhodium which got me interested in this process so thanks for that too. Any reason why one needs to have ALL the NaN3 dissolved? I mean could one safely attempt to use less DMSO than used in the given procedure?
(Stranger)
06-23-02 22:53
No 324649
I think I found another usefull article from the same place!
"Cobalt (II)Chloride-Catalyzed Chemoselective Sodium Borohydride reduction of azides in Water:
Reduction of azides to amines and amides was carried out with NaBH4/CoCl2 · 6 H2O in sole water at 25 °C under catalytic heterogeneous conditions. A broad spectrum of azides was reduced in a short time, chemoselectively in high yield and purity."
Synth, 2000 issue 5, p646
Theres even a proc to reuse the reducing agent and PTC(s)! All reported yields 90%+ in <= 20 min.!
http://www.thieme-connect.de
Imagine what someone who knows what they are doing might find in there?
(Master Whacker)
06-25-02 11:15
No 325198
(Rated as: excellent)
Convenient Procedure for One-pot Conversion of Azides to N-monomethylamine: Synlett 2001, 1003-1005
This reference is a significant advance in alkylazide reduction techniques because it reduces the azide and methylates it in one single step. This is the only ref I am aware of which yields a N-methylamine or N-ethylamine directly from the azide precursor without using catalytic hydrogenation for the reduction.
Sample procedure:
Secondary alkylazide(.148mmol) in CH2Cl2(1.5ml) was added a solution of (CH3)3P in toluene (1.0M, .3mL) at room temp. After stirring for 1.5hr, paraformaldehyde(22.6mg, 0.753mmol) was added. The rxn. mixture was stirred for an additional 6hr at room temp then rxn was cooled to 0'C and MeOH (2.0ml) and NaBH4(28mg, 0.74mmol) was added. After stirring for .5hr the rxn. was stopped with saturated aq. NaHCO3and extracted with 5 X 10ml CH2Cl2. Extracts were pooled and evaporate to yield 83% N-methylamine.
(Hive Bee)
06-25-02 19:27
No 325407
That sounds like a potentially very useful procedure...
Any idea if (Ph)3P could be used in place of (CH3)3P?
SpicyBrown
(Stranger)
06-25-02 22:28
No 325483
(Rated as: excellent)
Thanks for pulling that one up Ritter! I knew there was more good stuff there to be found. I guess I should stop being a lazy fucker and post the other procedures since some can not access them:
SYNTH OF ALKYL AZIDE
A stock solution of 0.5 M NaN3 in DMSO was prepared by stirring the solution for 24 hours at 25C. To a 100 ml round-bottom flask equipped with a magnetic stir bar, was added a 0.5M solution of NaN3 (0.715g,11mmol) in DMSO (22mL) at 25C. To this solution was added alkyl halide (10mmol), and the mixture was stirred untill all the starting material had been consumed, as observed by GC analyses. The reaction was quenched with H2O (50mL) [slightly exothermic] and stirred until it cooled to r.t. The reaction was extracted with Et2O(3x30mL); the Et2O extracts were washed with H2O (2x50mL) and once with brine (50mL). The organic layer was dried (MgSO4) filtered, and the solvent removed under vacuo(20 Torr) to afford the pure alkyl azide.
ALKYL AZIDE to AMINE
To a mixture of azide (2.0 mmol) and CoCl2*6H20 (0.048 g, 0.2mmol), and when necessary CTABr (0.07g o.2mmol) at 25C was added dropwise under stirring a solution of NaBH4 (0.152g, 4.0 mmol) in H2O (4mL). The formation of a black precipitate indicated the formation of a cobalt boride species. The mixture was stirred at 25C for more than 10min when necessary. At the end of the reaction the mixture was extracted with Et2O (5x10 mL). The organic phase was dried (NaSO4) and concentrated under reduced pressure to give the pure amine or amide.
Re-use of Reducing agent:
The pH of the remaining mother liquor (aprox 4ml) after extraction with Et2O of amine or amide was adjusted to 8.0 by adding a few drops of concd HCl. Azide (2.0mmol) was then added followed by NaBH4 (0.0152 g, 4.0 mmol) in small doses. Mixture was stirred at 25C for more than 10 min when necessary and then extracted with Et2O. The mother liquor can continue to be reused.
(PVC-Analog Taste-Tester)
07-08-02 21:41
No 330257
Why reduce with a borohydride or Al/Hg when calcium or Mg will get the job done. I'm assuming it's because the work-up's messy with either element, because Strike claimed near 100% yields when reducing?
Love my country, fear my government.
(Chief Bee)
07-09-02 00:16
No 330318
Bwiti: Noone has bother to test it yet, therefore it is not yet in the paradigm of how things are done. For two years noone bothered to test NaBH4 instead of NaBH3CN even though a suggested method existed - and then poof! Someone posts and says it actually works, and supplies a writeup. A few try it, and says it really does. A few months later, and it is the preveiling method.
We have too few bold researchers who test all the most important novel suggestions.
(Stranger)
07-10-02 03:20
No 330803
'What's wrong with Ca or Mg'
SW doesn't know he hasn't tried it. He is mostly interested in increasing the yields from ritters original idea. As far as he knows the hard part is getting good yields with NaN3 (?) He tried ritters 1 potter as is and also w aliquat 336, both gave relatively low yields although the workup was clean and easy. He thought the low yields were from the NaN3 stage. If one were using DMSO at room temp, how would one know when it's done? Should there be a color change? What color and density is bromo-azide anyways?
(Chief Bee)
04-02-04 09:49
No 498692
(Rated as: good read)
Versatile Reagent for Reduction of Azides to Amines
B. Pal, P. Jaisankar, V. S. Giri
Synthetic Communications 34(7), 1317-1323 (2004)
DOI:10.1081/SCC-120030322
Abstract
Triphenylphosphine in refluxing methanol effectively reduces a variety of azides to amines in very good yields.
Staudinger was the first to observe that TPP with azides form the phosphoazides, which spontaneously undergo nitrogen elimination resulting in iminophosphoranes. These iminophosphoranes on hydrolysis with water yield the corresponding amines and the TPP oxide. Drawbacks of Staudinger reaction being known, a few reagents were developed to expedite the reduction. All those methods require dual steps, i.e., the formation of iminophosphoranes followed by hydrolysis with water. The method developed by us spontaneously results in the formation of amines when the azide is treated with TPP/methanol. Moreover, the amines could be directly precipitated out from the crude reaction mixture as hydrochloride salts. In our case the iminophosphoranes resulting from reaction of TPP with azides undergo methanolysis to yield the corresponding amines and TPP oxide. A comparison of literature data for reduction of azides to amines using PPh3 reveals that TPP–MeOH is an ideal reagent for bringing about such a reduction. We also observed that the reduction takes place even if MeOH is replaced with EtOH or i-PrOH or n-BuOH, but MeOH appears to be a better solvent.
Reduction of Azides to Amines
Triphenylphosphine (1.5 mmol, 0.147 g) was added to a solution of azide (1 mmol) in dry methanol (10 mL). The reaction mixture was then refluxed at 80°C for 1 hr. After all the starting material had disappeared (monitored by TLC using 1:7 mixture of EtOAc–benzene solvent system), the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure to yield the crude amines.
The corresponding hydrochloride salts were obtained directly by the addition of few drops of hydrochloric acid to the solution of the crude reaction mixture in toluene (5 mL). The salts formed were filtered and recrystallized from chloroform–methanol (9:1) to afford the hydrochloride salts of the amines (85-94% yield).
Maybe someone could be kind enough to post the full paper?
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
04-03-04 02:42
No 498831
(Rated as: excellent)
Versatile Reagent for Reduction of Azides to Amines
B. Pal, P. Jaisankar, V. S. Giri
Synthetic Communications 34(7), 1317-1323(2004)
Abstract
Triphenylphosphine (TPP) in refluxing methanol effectively reduces a variety of azides 1a–k to amines 2a–k in very good yields.
Introduction
Azides1 have continued to attract the attention of synthetic organic chemists particularly because they could be utilized for preparing important compounds2, 3 containing nitrogen functionalities. Over the years a number of methodologies have been developed4 for reduction of azides to amines using variety of reagents.5
We have observed very recently6 that triphenylphosphine (TPP) in methanol could efficiently reduce maleimides to the corresponding succinimides in excellent yields. The reagent has since been used for reduction of azides 1a–k to amines 2a–k (Scheme 1). Herein, we report a simple and improved method for the reduction of azides 1a–k to the amines 2a–k using TPP in refluxing methanol for 1 hr and the results are shown in Table 1.
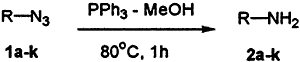
Scheme 1.
Table 1. Reduction of azides 1 to amines 2 by using PPh3-MeOH.
| 1 | Substrate | 2 | Product | Isolated Yield (%) |
| a | Benzyl azide | a | Benzylamine ∙ HCl | 93 |
| b | 4-Bromobenzyl azide | b | 4-Bromobenzylamine ∙ HCl | 93 |
| c | 4-Chlorobenzyl azide | c | 4-Chlorobenzylamine ∙ HCl | 92 |
| d | 4-Fluorobenzyl azide | d | 4-Fluorobenzylamine ∙ HCl | 93 |
| e | 3-(2-Ethyl-azido)-indole | e | Tryptamine ∙ HCl | 94 |
| f | 1-Azido-2,4-dinitrobenzene | f | 2,4-Dinitroaniline ∙ HCl | 85 |
| g | 3-Azido-1,2:5,6-di-O-cyclohexylidene-3-d |
g | 3-Amino-1,2 5,6-di-O-cyclohexylidene-3-deoxy-á-D-all |
85 |
| h | Benzoyl azide | h | Benzamide | 80 |
| i | 3-Pyridinecarboxylic acid azide | i | Nicotinamide | 80 |
| j | 4-Methylbenzenesulfonyl azide | j | (4-Methyl)-benzene sulfonamide | 82 |
| k | Methansulfonyl azide | k | Methansulfonamide | 81 |
Staudinger and Meyer7 were the first to observe that TPP with azides form the phosphoazides, which spontaneously undergo nitrogen elimination resulting in iminophosphoranes. These iminophosphoranes on hydrolysis with water yield the corresponding amines and the TPP oxide. Drawbacks of Staudinger reaction being known, a few reagents were developed to expedite the reduction. Charette et al.8 have used resin bound TPP to reduce azides to amines but this requires prolonged heating of iminophosphoranes with water for long hours (36 hr). A solution phase parallel synthesis developed by Lindsley et al.9 is no doubt an improvement but involves the use of costly fluorous-tethered-TPP. All the above-mentioned methods require dual steps, i.e., the formation of iminophosphoranes followed by hydrolysis with water. The method developed by us spontaneously results in the formation of amines when the azide is treated with TPP/methanol. Moreover, the amines 2a–e could be directly precipitated out from the crude reaction mixture as hydrochloride salts. On the other hand, the amides resulting from reduction of acid azides were purified by column chromatography over silica gel. In our case the iminophosphoranes resulting from reaction of TPP with azides undergo methanolysis to yield the corresponding amines and TPP oxide. The TPP oxide has been isolated from the reaction mixture and confirmed by m.p., mmp, and 31P NMR. A comparison of literature data for reduction of azides to amines using PPh3 reveals that TPP–MeOH is an ideal reagent for bringing about such a reduction. We also observed that the reduction takes place even if MeOH is replaced with EtOH or i-PrOH or n-BuOH, but MeOH appears to be a better solvent (Table 2)
Table 2. Solvent effect on reduction of azide 1a to amine 2a.
| Solvent | Time | Isolated Yield (%) |
| MeOH | 1 h | 93 |
| EtOH | 1 h 15 min | 89 |
| i-PrOH | 1 h 15 min | 90 |
| n-BuOH | 1 h 30 min | 85 |
All the products are known in the literature and have been characterized by direct comparison with authentic samples prepared by classical methods. The azides were obtained by following standard procedures10, 11 and characterized from their physical data. The azides exhibit characteristic IR absorbance band between 2090 and 2140 cm-1.
Melting points were determined in open capillaries and are uncorrected. IR spectra were recorded in FT/IR 410 JASCO spectrophotometer. 1H NMR and 13C NMR spectra were measured in a 300 MHz BRUKER 300 DPX spectrophotometer using TMS as internal standard. The mass spectra were run on a JEOL AX-500 spectrometer at 70 eV. Elemental analyses were carried out on a Perkin Elmer 2400 Series II CHNS/O analyzer. Optical rotations were recorded at 25°C in P-1020 JASCO polarimeter. Petroleum ether refers to the fraction boiling in the range 60-80°C. Solvents like petroleum ether, chloroform, methanol, ether, THF, DMF, and DMSO were purified and dried before use. Column chromatography was carried out on silica gel of 60–120 mesh. TLC was done on standard Merck TLC aluminum sheets. Unless otherwise mentioned the compounds have been crystallized from either petroleum ether-CHCl3 mixture or MeOH.
Typical Procedure for Reduction of Azides 1a–k to Amines 2a–k
TTP (1.5 mmol, 0.147 g) was added to a solution of azide (1 mmol) in dry methanol (10 mL). The reaction mixture was then refluxed at 80°C for 1 h. After all the starting material had disappeared (monitored by TLC using 1:7 mixture of EtOAc-benzene solvent system), the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure to yield the crude amines/amides. In case of amines, the corresponding hydrochloride salts were obtained directly by the addition of few drops of hydrochloric acid to the solution of the crude reaction mixture in toluene (5 mL). The salts formed were filtered and recrystallized from chloroform-methanol (9:1) to afford the hydrochloride salts of the amines 2a–e. The amines 2f, 2g, and the amides 2h–k were obtained by evaporating the solvent from the reaction mixture followed by chromatography of the residue over silica gel (60-120 mesh). Elution with petroleum ether-chloroform (1:3) gave TPP oxide as white solid. Further elution with chloroform–methanol (50:1) solvent mixture yielded the pure amides/amines.
Benzyl azide (1a). B.p. 98°C/16 mm Hg (lit.12 b.p. 108°C/23 mm Hg).
4-Bromobenzyl azide (1b). IR (Neat): 2922, 2099, 1702, 1589, 1486 cm-1; 1H NMR (CDCl3): ä4.30 (s, 2H), 7.20 (d, J = 6.7 Hz, 2H), 7.52 (d, J = 6.5 Hz, 2H). m/z: 213 (M + 2, 20), 211 (M+, 18), 171 (98), 169 (100), 157 (38), 155 (35). Anal. calcd for C7H6BrN3: C, 39.65; H, 2.85; N, 19.82. Found: C, 39.56; H, 2.91; N, 19.89.
4-Chlorobenzyl azide (1c). IR (Neat): 3031, 2096, 1592, 1442, 1257, 1192 cm-1; 1H NMR (DMSO-d6): ä3.40 (s, 2H), 7.40 (d, J = 8.4 Hz, 2H), 8.09 (d, J = 8.6 Hz, 2H); m/z: 167 (M+, 30), 125 (100), 112 (35). Anal. calcd for C7H6ClN3: C, 50.17; H, 3.61; N, 25.07. Found: C, 50.09; H, 3.68; N, 25.15.
4-Fluorobenzyl azide (1d). IR (Neat): 2099, 1391, 1108 cm-1; 1H NMR (CDCl3): ä3.93 (s, 2H), 7.40–7.48 (m, 2H), 8.02–8.08 (m, 2H); m/z: 151 (M+, 35), 109 (95), 96 (39). Anal. calcd for C7H6FN3: C, 55.63; H, 4.00; N, 27.80. Found: C, 55.47; H, 4.08; N, 27.96.
3-(2-Ethyl-azido)-indole (1e). IR (Neat): 3410, 2924, 2098, 1455, 1259, 1097 cm-1; 1H NMR (CDCl3): ä3.03 (t, J = 6.8 Hz, 2H), 3.52 (t, J = 6.8 Hz, 2H), 6.98 (s, 1H), 7.09–7.22 (m, 2H), 7.30 (d, J = 7.7 Hz, 1H), 7.57 (d, J = 7.4 Hz, 1H), 7.92 (brs, 1H, NH); m/z: 186 (M+, 56), 144 (100). Anal. calcd for C10H10N4: C, 64.50; H, 5.41; N, 30.09. Found: C, 65.47; H, 5.48; N, 30.17
1-Azido-2,4-dinitrobenzene (1f). M.p. 69–71°C (lit.17 m.p. 65–68°C).
3-Azido-1,2:5,6-di-O-cyclohexylidene-3-deoxy-á-D-allofuranos
Benzoyl azide (1h). M.p. 34–35°C (lit.20 m.p. 32°C); IR (Neat): 2139, 1711, 1227 cm-1; 1H NMR (CDCl3): ä7.40–7.52 (m, 3H), 7.58–7.65 (m, 1), 8.10–8.15 (m, 1H); m/z: 147 (M+, 70), 105 (100), 77 (90).
3-Pyridinecarboxylic acid azide (1i). M.p. 49–50°C (lit. m.p. 47–48°C).
4-Methylbenzenesulfonyl azide (1j). M.p. 24–26°C (lit. m.p. 22°C).
Benzylamine • HCl (2a). M.p. 258–260°C (lit.13 m.p. 255–257°C).
4-Bromobenzylamine • HCl (2b). M.p. 258–260°C (lit.14 m.p. 279°C).
4-Chlorobenzylamine • HCl (2c). M.p. 260–262°C (lit.15 m.p. 259°C).
4-Fluorobenzylamine • HCl (2d). M.p. 282–284°C; IR (KBr): 3014, 1592, 1514, 1239 cm-1; 1H NMR (DMSO-d6): ä4.00 (brs, 2H), 7.20–7.28 (m, 2H), 7.53–7.60 (m, 2H), 8.54 (brs, 2H, NH2); m/z: 125 (M+, 43). Anal. calcd for C7H8FN: C, 67.18; H, 6.44; N, 11.19. Found: C, 67.08; H, 6.53; N, 11.24.
Tryptamine • HCl (2e). M.p. 250–252°C (lit.16 m.p. 248°C).
2,4-Dinitroaniline • HCl (2f). M.p. 172–174°C (lit.18 m.p. 180°C).
3-Amino-1,2:5,6-di-O-cyclohexylidene-3-deoxy-á-D-allofuranos
Benzamide (2h). M.p. 126–128°C (lit.21 m.p. 130°C).
Nicotinamide (2i). M.p. 125–127°C (lit.23 m.p. 129-130°C).
(4-Methyl)-benzene sulfonamide (2j). M.p. 134–136°C (lit.25 m.p. 138°C).
Methansulfonamide (2k). M.p. 84–86°C (lit.26 m.p. 88°C
References
[1] Scriven E. F.V., Turnbull K., Chem. Rev., 88 (1988) 297.
[2] Smith S. C., Heathcock C.-H., J. Org. Chem., 57 (1992) 6379.
[3] McDonald F. E., Danishefsky S. J., J. Org. Chem., 57 (1992) 7001.
[4a] Kamal A., Ventata Ramana K., Ankati H. B., Venkata Ramana A., Tetrahedron Lett., 43 (2002) 6861. and references cited therein.
[4b] Ito M., Koyakumaru K., Takaya T. H., Synthesis, (1995) 376.
[4c] Molina P., Diaz I., Tarraga A., Tetrahedron, 51 (1995) 5617.
[4d] Ramesha A. R., Bhat S., Chandrasekaran S., J. Org. Chem., 60 (1995) 7682.
[4e] Capperucci A., Degl'Innocenti A., Funicello M., Mauriello G., Scafato P., Spagnolo P., J. Org. Chem., 60 (1995) 2254.
[4f] Kamal A., Reddy B. S.P., Reddy B. S.N., Tetrahedron Lett., 37 (1996) 6803.
[4g] Huang Y., Zhang Y., Wang Y., Tetrahedron, 38 (1997) 1065.
[4h] Said B., Thierry L. G., Michel V., Rene G., Loic T., Robert C., Tetrahedron Lett., 28 (1987) 1761.
[4i] Srinivasan N., Ganem B., J. Org. Chem., 52 (1987) 5044.
[5] Molander G. A., Chem. Rev., 92 (1992) 29.
[6] Pal B., Pradhan P. K., Jaisankar P., Giri V. S., Synthesis, 10 (2003) 1549.
[7] Staudinger H., Meyer J., Helv. Chim. Acta, 2 (1919) 635.
[8] Charette A. B., Bozei A. A., Janes M. K., Org. Lett., 2 (2000) 3777.
[9] Lindsley C. W., Zhao Z., Newton R. C., Leister W. H., Strauss K. A., Tetrahedron Lett., 43 (2002) 4467. and references cited therein.
[10] Boyer J. H., Mack C. H., Goebel N., Morgan L. R., J. Org. Chem., 23 (1958) 1051.
[11] Marco-Contelles J., Fernandez M. R., J. Org. Chem., 66 (2001) 3717.
[12] Curtius E., Ber., 55 (1922) 1559.
[13] Egli R. A., Helv. Chim. Acta, 53 (1970) 47.
[14] Rupe H., Helv. Chim. Acta, 13 (1930) 466.
[15] Feuer H., Braunstein D. M., J. Org. Chem., 34 (1969) 1817.
[16] Jackson A. H., Smith A. E., J. Chem. Soc., (1965) 3498.
[17] Okamoto T., Hayashi S., Yakuga. Zasshi, 86 (1966) 766.
[18] Ashton A. A., J. Chem. Educ., 40 (1963) 545.
[19] Collins P. M. Carbohydrates, Chapman and Hall Ltd., 1987, p. 136.
[20] Bergel Z., Angew. Chem., 40 (1927) 974.
[21] Nishinaga A., Shimizu T., Matsuura T., J. Chem. Soc., Chem. Commun., (1979) 970.
[22] Paraskewas S., Synthesis, (1974) 819.
[23] Forge L. F.B., J. Am. Chem. Soc., 50 (1928) 2480.
[24] Doering W. V.E., DePuy C. H., J. Am. Chem. Soc., 75 (1953) 5955.
[25] Heillbron I., Bunbury H. M. Dictionary of Organic Compounds, Oxford University Press, 1953, p. 517.
[26] Billeter O. C., Ber., 38 (1905) 2015.