(Stranger)
04-30-03 20:23
No 430441
(Rated as: excellent)
J. Org. Chem., 1999, 64, 8962-8964
A simple method for the reduction of carboxylic acids to aldehydes or alcohols using H2 and Pd/C
Aldehydes are versatile compounds in organic synthesis. Despite their intrinsic benefits, there are relatively few methods for their preparation.1 A common approach to obtain aldehydes is in fact the oxidation of primary alcohols2 or the reduction of carboxylic acids and their derivatives.3 This last transformation is particularly useful for the preparation of N-protected R-amino aldehydes4 that are valuable intermediates for the synthesis of biologically active compounds.5 Several methods employed for the preparation of protected amino aldehydes
make use of complex metal hydrides as the reducing agents and esters or amides as the starting material (for example DIBAL-H on methyl esters6 and LiAlH4 on particularly reactive amides7).
All these approaches suffer from several disadvantages:
metal hydrides are generally highly reactive or expensive, some problems may occur during the separation of the aldehyde from the reaction mixture after the quench of the metal hydride, and the reaction conditions can produce sometimes racemization of the formed aldehyde.
Consequently, a practicable alternative for the large-scale preparation of amino aldehydes is the reduction of the carboxylic acid to alcohol followed by reoxidation of the alcohol to aldehyde.8 Except for the classical Rosenmund catalytic hydrogenation of acyl chlorides and some related reactions,9 few reports describe the direct reduction of carboxylic acid and their derivatives using hydrogen and a catalyst.10
For example, in a Rosenmund type reduction a couple of the major problems are the stereochemical lability of the acyl chlorides and the difficulties in their preparation in the presence of acid labile groups or protections.11
Following our interest in the use of [1,3,5]triazine derivatives in organic synthesis,12 we discovered that the activated ester of N-Boc amino acids with 2-chloro-4,6-dimethoxy[1,3,5]triazine (1) can be reduced into the corresponding aldehydes in good yields with H2 on catalytic Pd/C.
Treating a carboxylic acid 3 with the complex formed by 2-chloro-4,6-dimethoxy[1,3,5]triazine (1) and N-methylmorpholine in DME (adduct 2 in Scheme 1), the corresponding activated ester 4 is quantitatively formed.13 A solution of this ester can be directly treated with H2 (1 atm pressure) at room temperature in the presence of Pd catalyst (Pd/C 10%) to give the corresponding aldehyde 5. After filtration of the catalyst and acidic work up to separate the [1,3,5]triazine byproducts, the aldehyde can be recovered practically pure by evaporation of the solvent or purified by standard methods. Consequently, aldehydes are obtained from carboxylic acids using a catalytic process and friendly reaction conditions.
This process, although simple, will require optimization of the solvent, reaction time, and H2 pressure. The best results for conversion and isolated yields of the aldehyde were obtained using DME or THF in the formation of the activated ester and EtOH in the reductive step.
On the other hand, when the reaction was carried out for longer times, the alcohol obtained from over-reduction of the aldehyde was the main component of the reaction mixture. For example, in the case of the activated ester formed from acid 3i, after 3 h of reduction at room temperature, we observed the formation of the aldehyde 5i with a conversion of about 85%. On waiting one additional hour for the complete disappearance of the activated ester, we observed the formation of the corresponding alcohol 6i that, after additional 4 h of reduction, became the predominant product. Nevertheless, as alcohol does not react with 4 in the absence of a proper activation,14 we did not observe the formation of the symmetric ester as often happens in Rosenmund type reductions.15
The formation of the alcohol was also observed upon increasing the H2 pressure. When the reaction was carried out in a Parr apparatus (3-5 atm of H2, room temperature) on compounds 3f,j-i, the corresponding alcohols 6f,j-i were obtained in high yield (Scheme 2). The direct conversion of a carboxylic acid into the corresponding alcohol by catalytic hydrogenation was thus achieved.
This methodology was applied to aliphatic acids and N-Boc-amino acids. When the reaction was carried out on aromatic carboxylic acids (3e,f), the main reaction product was the alcohol, even at low temperature and H2 pressure. The use of a poisoned Pd catalyst did not improve the yields of the aldehyde.16
Nevertheless, as the intermediate ester 4 is not consumed by the alcohol formed during the reaction, we were able to isolate a limited amount of the aldehyde at the end of the reaction. The formation of the alcohol may happen also during the reduction of N-Boc amino acids. We observed that even in the case of similar substrates, different reaction times were needed for a satisfactory conversion to aldehydes. For example, the transformation of N-Boc-valine (3l) occurred after 3 h of reduction at room temperature, whereas N-Boc-leucine (3i) could be successfully reduced to aldehyde 5i exclusively at 0 °C for 2 h. For this reason we suggest trying this procedure first on small amounts of the substrate to find the correct conditions before attempting the reduction of the whole material of interest. We also recommend use of a low pressure of H2 (using an H2 buret or a balloon) and eventually carrying out the reaction at 0 °C to prevent the formation of the alcohol. On the other hand, if the alcohol is desired, the reduction under 5 atm of H2 gives quantitative yields of the products.
Significant racemization of R-amino aldehydes did not occur under these conditions, as revealed by the optical rotation values of the products and their derivatives (semicarbazones and dinitrophenyl hydrazones; see the Supporting Information).
We think that the method described in this note can be very useful for the preparation of aldehydes (and alcohols) in large scale, as it does not use anhydrous solvents, strong reaction conditions, or dangerous or expensive reagents. The main drawback (in the case of the aldehyde) is the necessity to monitor the reaction to prevent the formation of the alcohol, but we are currently trying to improve the conditions to avoid this trouble also.
Experimental Section
All the solvents and the reagents (including Pd/C) were used in the commercially available grade purity. The protected amino acids were purchased and their purity was established before utilization by melting point and optical rotation. Although 2-chloro-4,6-dimethoxy[1,3,5]triazine is commercially available, we prepared it following a published procedure.17
General procedure for the reduction of carboxylic acids to aldehydes:
(S)-2-(tert-Butoxycarbonylamino)-propana
The ethereal layer was separated and dried over anhydrous Na2SO4 to give, after evaporation of the solvent, product 5h (0.80 g, 79% yield) practically pure for any further transformation of the aldehyde group. An analytical sample was purified by column chromatography on silica gel.
The identity of the product was determined by comparison of 1H NMR data, [R] values, and semicarbazone melting point with those previously described in the literature.18 1H NMR (300 MHz, CDCl3) ‰ 9.53 (s-like, 1H), 5.05 (bs, 1H), 4.2 (m, 6 components visible, 1H), 1.42 (s, 9H), 1.32 (d, J ) 7 Hz, 3H). [R]D ) -25.0 (c ) 1, MeOH); lit.18 [R]D ) -25.52 (c ) 1, MeOH). Semicarbazone: [R]D ) -24.3 (c ) 1, MeOH); lit.18 [R]D ) -23.82 (c ) 1, MeOH). Anal. Calcd for C9H18N4O3 (250.17): C, 46.95; H, 7.87; N, 24.34. Found: C, 46.77; H, 7.88; N, 24.30.
General Procedure for the Catalytic Reduction of Carboxylic Acids to Alcohols.
(S)-2-(tert-Butoxycarbonylamino)-3-pheny
The identity of the product was determined by comparison of melting point, 1H NMR data and the [R] values with those previously described in the literature.19,20 6j: mp 94-95 °C; lit.19 mp 94-96 °C; [R]D ) -20 (c ) 1, MeOH); lit.20 [R]D ) -21.6. 1H NMR (300 MHz, CDCl3) ‰ 7.2 (m, 5H), 5.0 (bs, 1H), 4.4 (m, 4 components visible, 1H), 4.2 (m, 2H), 2.2 (m, 2H), 2.0 (bs, 1H), 1.45 (s, 9H).
(1) Larock, R. C. Comprehensive Organic Transformation; VCH Publisher: New York, 1989; p 585.
(2) Epstein, W. W.; Sweat, F. W. Chem. Rev. 1967, 67, 247.
(3) Cha J. S. Org. Prep. Proced. Int. 1989, 21, 451.
(4) Jurczak, J.; Golebiowski, A. Chem. Rev. 1989, 89, 149.
(5) For a series of recent applications, see: Kim, D. H.; Jin, Y. Bioorg. Med. Chem. Lett. 1999, 9, 691. Han, Y.; Chorev. M. J. Org. Chem. 1999, 64, 1972. Salvino, J. M.; Mervic, M.; Mason, H. J.; Kiesov, T.; Teager, D.; Airey, J.; Labaudiniere, R. J. Org. Chem. 1999, 64, 1823. Wrodnigg, T. M.; Stu¨z, A. E. Angew. Chem., Int. Ed. Engl. 1999, 38, 827. Steuer, S.; Podlech, J. Eur. J. Org. Chem. 1999, 1551. Gennari C.; Longari C.; Ressel S.; Salom B.; Mielgo A. Eur. J. Org. Chem. 1998, 945, 5. Bonini B. F.; Comes-Franchini, M.; Fochi, M.; Gawronski, J.; Mazzanti, G.; Ricci, A.; Varchi, G. Eur. J. Org. Chem. 1999, 437. Kokotos, G.; Padron, J. M.; Martin, T.; Gibbons, W. A.; Martin, V. S. J. Org. Chem. 1998, 63, 3741. Armbruster, J.; Grabowski S.; Ruch, T.; Prinzbach H. Angew. Chem., Int. Ed. Engl. 1998, 37, 2242.
(6) Luly, J. R.; Hsiao, C.-N.; Bamung, N.; Plattner, J. J. J. Org. Chem. 1988, 53, 6109. Garner, P.; Park, J. M. J. Org. Chem. 1987, 52, 2361.
(7) Campbell, A. D.; Raynham, T. M.; Taylor, R. J. K. Synthesis 1998, 1707. Godjoian, G.; Singaram, B. Tetrahedron Lett. 1997, 38, 1717. Fehrentz, J. A.; Castro, B. Synthesis 1983, 676.
(8) Dondoni, A.; Perrone, D. Synthesis 1997, 527. Konradi, A. W.; Kemp, S. J.; Pedersen, S. F. J. Am. Chem. Soc. 1994, 116, 1316. Leann, M. R.; Sowin, T. J.; Morton, H. E. Tetrahedron Lett. 1992, 33, 5029. Leanna, M. R.; Sowin, T. J.; Morton, H. E. Tetrahedron Lett. 1992, 33, 5029. Rodriguez, M.; Llinares, M.; Doulut, S.; Heitz, A. Martinez, J. Tetrahedron Lett. 1991, 32, 923. Anelli, P. L.; Biffi, C.; Montanari, F.; Quici, S. J. Org. Chem. 1987, 52, 2559.
(9) Babler, J. H.; Invergo, B. J. Tetrahedron Lett. 1981, 22, 11. Four, P. Guibe, F. J. Org. Chem. 1981, 46, 4439. Fleet, G. W. J.; Harding, P. J. C. Tetrahedron Lett. 1979, 20, 975. Burgstahler, A. W.; Weigel, L. O.; Shaefer, C. G. Synthesis, 1976, 767. Citron, J. D. J. Org. Chem. 1969, 34, 1977.
(10) Braden, R.; Himmler, T. J. Organomet. Chem. 1989, 367, C12. For alternative approaches, see: Chandrasekhar, S.; Suresh Kumar, M.; Muralidhar, B. Tetrahedron Lett. 1998, 39, 909. Shamsuddin, K. M.; Zobairi, M. O.; Musharraf, M. O. Tetrahedron Lett. 1998, 39, 8153. Ho, P. T.; Ngu, K-y. J. Org. Chem. 1993, 58, 2313. Fukuyama, T.; Lin, S.-C.; Li, L. J. Am. Chem. Soc. 1990, 112, 7050. Muraki, M. Mukaijama, T. Chem. Lett. 1974, 1447.
(11) See for example: Carpino, L. A.; Mansour, E.-S. M. E.; Sadat- Aalaee, S. Y. J. Org. Chem. 1991, 56, 2611. Carpino, L. A.; Cohen, B. J.; Stephens, K. E., Jr; Sadat-Aalee, S. Y.; Tien, J.-H.; Langridge, D. C. J. Am. Chem. Soc. 1986, 51, 3734.
(12) Falorni, M.; Porcheddu, A. Taddei, M. Tetrahedron Lett. 1999, 40, 4395.
(13) Kaminski, Z. J.; Paneth, P.; Rudzinki, J. J. Org. Chem. 1998, 63, 4248.
(14) Kaminska, J. E.; Kaminski, Z. J.; Gora, J. Synthesis 1999, 593.
(15) Rosenmund, K. W. Chem. Ber. 1918, 51, 585. For a critical discussion on Rosenmund reduction, see: Mossettig, E.; Mozingo, R. Org. Reactions 1948, 4, 362.
(16) We tried only Pd-BaSO4 5% and not Pd-C quinoline-S catalyst as described: Rachlin, A. I.; Gurien, H.; Wagner, D. P. Org. Synth. 1971, 51, 8.
(17) Cronin, J. S.; Ginah, F. O.; Murray, A. R.; Copp, J. D.Synth. Commun. 1996, 26, 3491.
(18) Stanfield, C. F.; Parker, J.; Kanellis, P. J. Org. Chem. 1981, 46, 4797. Hamada, Y.; Shioiri, T.; Chem. Pharm. Bull 1982, 30, 1921. Fehrentz, J. A.; Pothion, C.; Califano, J. C.; Coffet, A.; Martinez, J. Tetrahedron Lett. 1995, 35, 9031. Zlatoidsky, P. Helv. Chim. Acta 1994, 77, 545. Zlatoidsky P. Tetrahedron Lett. 1995, 36, 7281
(19) Rubini, E.; Gilon, C.; Selinger, Z.; Choreu, M. Tetrahedron 1986, 42, 6032.
(20) Kokotos, G. Synthesis 1990, 299.
Comment by Lego: The authors reduce only two benzoic acids, one is benzoic acid itself, the other is phtalic acid mono methylester. The yields are 32% and 30%. Cinnamic acid is reduced with a 76% yield. Generally it seems that aliphatic acids are easier to reduce with this method (phenylpropionic acid, 79%), best yield is stearic acid with 88%. But nonetheless this methods offers a new possibility.
The candle that burns twice as bright burns half as long
(Hive Bee)
06-11-03 19:55
No 439387
A very good find, there has been much interest in the reduction of amino carboxylic acids over the years. I've read and followed many reports and approches , some needing NaBH4/I , H2SO4, while others cyclohexanone, Tetanocene, and the list goes on. It has been my contention that COOH can be reduced to and alcohol through Hydrogenation and a catalyst. In this method, although it still needs to go through some hoops and jumps, gets you there but the research is old 1998 or older. There is some work being done where maybe a catalyst complex of palladium and silane may not only do the reduction but all the way to an alkane.
So congratulations for your find and the search will continue...java
We're all in this world together,
http://www.chiapaslink.ukgateway.net/
(Moderator)
10-13-03 18:04
No 464427
Check out Post 464426 (Lilienthal: "acid chlorides to aldehydes with formic acid", Tryptamine Chemistry) for a catlyst-free reduction.
(Hive Bee)
06-14-04 20:28
No 513353
Lego , Here is a related approach,.......java
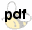
Brief....Recently, 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) has been described as a versatile reagent for the selective formation of amides1. Although CDMT has been shown to be a useful non-carbodiimide reagent, in terms of stability, mild reaction conditions and cost, it has the disadvantage to be irritating to the eye and nose. In addition, reactions are generally conducted under dry conditions by a two-step procedure: the carboxylic acid is first treated with CDMT in the presence of N-methylmorpholine to activate the acid, then an amine or an alcohol is added to obtain the product. This requires confirmation of the completion of the first step for successful results. To resolve these issues, a more convenient one-step approach is desired. In 1999 Kunishima et. al. reported the synthesis2 and use of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-m
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/
(Hive Bee)
06-17-04 03:13
No 513869
(Rated as: good read)
Chem. Pharm. Bull. 50(4) 549—550 (2002) 549
A Racemization Test in Peptide Synthesis Using 4-(4,6-Dimethoxy-1,3,5-
triazin-2-yl)-4-methylmorpholinium Chloride (DMT-MM)
[Munetaka KUNISHIMA,*,a,b Akiko KITAO,a Chiho KAWACHI,a Yasunobu WATANABE,a Shin IGUCHI,a,b
Kazuhito HIOKI,a and Shohei TANIa,b[/i]
Abstract Racemization of the C-terminal amino acid (Ala) has been studied in various solvents during coupling
between 4-methoxybenzyloxycarbonyl (Z(OMe))-Gly-L-Ala-OH and phenylalanine benzyl ester (H-Phe-OBzl)
with 4-(4,6-dimethoxy-1,3,5-thiazin-2-yl)-4-m
without substantial racemization in AcOEt, tetrahydrofuran (THF), N,N-dimethylformamide (DMF), CH3CN,
and 2-PrOH, while a slight racemization was observed in dimethyl sulfoxide (DMSO), EtOH, and MeOH. The
extent of racemization may correlate with the polarity of the solvents.
Excerp..... In addition, it is noteworthythat DMT-MM enables us to carry out the direct one pot condensation of carboxylic acids and amines in protic solvents like methanol, ethanol, 2-propanol, or water, as well as in a variety of aprotic organic solvents.
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/
(Hive Bee)
06-21-04 22:41
No 514657
Just so that everything is kept in a continous file , here are the synthesis of DMT-MM and CMDT as provided by Kinetic
Post 514650 (Kinetic: "DMT-MM articles", Chemistry Discourse)
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/
(Hive Bee)
10-09-04 02:54
No 534988
Synthesis of aromatic aldehydes via NiCl2 reduction and hydrolysis of oxazolines
M. Suresh Babu and K.M. Lokanatha Rai
Tet. Lett., 2004, 45(42) , 7969-7970
DOI:10.1016/j.tetlet.2004.07.119
Abstract: Reduction of 2-aryl-oxazolines with NiCl2/NaBH4 followed by hydrolysis gives the corresponding aldehydes in good yields.
Benzoic acid was converted into the corresponding oxazoline using a literature method.11 The oxazoline was treated with NiCl2/NaBH4 at -10 °C followed by hydrolysis with 5% aq HCl to give the corresponding aldehyde in 90% yield. This encouraging result prompted us to study some other commercially available aromatic acids and indeed excellent yields of the corresponding aldehydes were obtained (Table 1). Unfortunately reduction of the oxazoline derived from propanoic acid under identical conditions gave only a very low yield of propanal. All the products prepared were identical by bp or mp and by IR and NMR spectroscopy with known aldehydes.12
General procedure for the preparation of benzaldehyde from benzoic acid
In the first step of the reaction, 2-phenyl-2-oxazoline (5 mmol, 0.735 g) and NiCl2 (2.37 g, 10 mmol) were taken in methanol and cooled to -10 °C. Sodium borohydride (0.74 g, 20 mmol) was added in small amounts over 30 min whilst maintaining the temperature at -10 °C. The mixture was then kept at 10 °C for a further 30 min to complete the reaction. The reaction mixture was filtered through Celite and the solvent evaporated under vacuum. The residue was treated with 5% HCl (10 mL) at 40 °C for 10-15 min, then cooled to room temperature and extracted with diethyl ether (25 mL). The organic layer was washed with water, dried over anhydrous sodium sulfate and evaporated under vacuum to give benzaldehyde as a colourless oily liquid in 90% yield (0.477 g).
11. Wiley, R. H.; Bennett, L. L., Jr. Chem. Rev. 1949, 44, 447–476 [Refluxing hydroxyamides (ArCONHCH2CH2OH) with SOCl2 followed by treatment with Na2CO3 gave the oxazolines derivatives in typically 65% overall yield; Fry E. M. J. Org. Chem. 1949, 14, 887–894].
12. Furniss, B. S.; Hannaford, A. J.; Smith, P. W. G.; Tatchell, A. R. In Vogel’s Text Book of Practical Organic Chemistry, 5th ed.; 1994; pp 1334–1335.
The chemistry of the oxazolines
Wiley, R. H., Bennett, L. L.
Chem. Rev., 1949, 44, 447–476
Oxazolines
Fry E. M.
J. Org. Chem., 1949, 14, 887–894
Ethanolamine was benzoylated in sodium bicarbonate solution at 15°. The product was taken into chloroform, recovered, and distilled at 179-189° (1 mm.). The crude product (85% yield) was recrystallized by dissolving in warm ethyl acetate, then adding dry ether; m.p. 61-63', 64% yield.
1-Phenyloxazoline hydrochloride (III-HCl). beta-Hydroxyethylbenzamide (2.0 g.) was added portionwise to 4 ml of chilled thionyl chloride at not over 13°. The solution was kept in the ice-bath for one hour; excess thionyl chloride was then removed under reduced pressure and the crystalline product brought to constant weight (2.3 g.) while keeping cold. The theoretical weight for the oxazoline hydrochloride is 2.22 g. The salt melted at 101-103° with a slight sinter at 77°. It was decomposed with sodium carbonate solution, the base removed with ether and the carbonate solution analyzed for sulfite and chloride ions. The ratio of the moles of sulfur dioxide to that of starting material was 0.03 to 1.0. Chloride was found in small excess, 105 % of theory. As reported (4) the oxazoline hydrochloride is stable in solution at room temperature. Recrystallization was effected by adding acetone to an aqueous solution; m.p. 75-76°. The value previously given (4) is 80-81°, and it is possible the lower melting point and low analytical value are due t o loss of hydrogen chloride. The phenomenon of slight sintering at the melting point with transformation into p-chloro-ethylbenzamide, m.p. 101-103°, (see above) was observed solely with the crude reaction product.
The tendency is to push it as far as you can
(Hive Bee)
10-18-04 19:00
No 536384
(Rated as: excellent)
Facile Reduction of Carboxylic Acids, Esters, Acid Chlorides, Amides and Nitriles to Alcohols or Amines Using NaBH4/BF3•Et2O
Su-Dong Cho, Yong-Dae Park, Jeum-Jong Kim, J. R. Falck, and Yong-Jin Yoon
Bull. Korean Chem. Soc. 2004, 25(3), 407-409
General Procedure: Reduction of Carboxylic acids and Carboxylate Derivatives.
A solution of BF3•Et2O (0.0065 mmol) in THF (15 mL) was added slowly to a room temperature solution of NaBH4 (0.015 mmol) and carboxylic acids or carboxylate derivatives (0.01 mmol) in THF (25 mL) under an inert atmosphere. The mixture was heated to reflux until TLC monitoring showed complete consumption of the substrate. The reaction mixture was cooled to 0°C, quenched with water (caution: vigorous gas evolution) keeping the temperature at 10°C. After 10 min, the THF was removed under reduced pressure, CH2Cl2 (or Et2O) was added, and the stirring was continued for another 1 h. The organic layer was separated, washed with brine, dried over MgSO4, and the solvent was removed under reduced pressure. Purification of the residue by SiO2 chromatography gave pure alcohol in the indicated yield (Table 1).
Table 1. NaBH4/BF3•Et2O reductions of carboxylic acids, esters, acid chloride, amides and nitrile.
| Entry | Substrate | Product | Time (h) | Yield (%) |
| 1 | Benzoic acid | Benzyl alcohol | 1 | 97 |
| 2 | 4-Methylbenzoic acid | 4-Methylbenzyl alcohol | 1 | 95 |
| 3 | Salicylic acid | Salicyl alcohol | 1 | 93 |
| 4 | 4-Nitrobenzoic acid | 4-Nitrobenzyl alcohol | 5 | 94 |
| 5 | 4-Chlorobenzoic acid | 4-Chlorobenzyl alcohol | 1 | 95 |
| 6 | Nicotinic acid | Nicotinyl alcohol | 0.5 | 94 |
| 7 | 5-Nitro-2-furoic acid | 5-Nitrofurfuryl alcohol | 1.5 | 98 |
| 8 | 4-Methoxybenzoic acid | 4-Methoxyphenethyl alcohol | 0.5 | 96 |
| 9 | Cyclohexanoic acid | Cyclohexylmethanol | 1 | 97 |
| 10 | Mucochloric acid | 2,3-dichloro-5H-furan- 2-one |
1.5 | 87a |
| 11 | (S)-(+)-Ethyl lactate | (S)-(+)-1,2-Propanediol | 3 | 94 |
| 12 | Ethyl 4-chloroacetoacetate | 4-Chlorobutanol | 1.5 | 76 |
| 13 | Benzoyl chloride | Benzyl alcohol | 1 | 96 |
| 14 | Benzamide | Benzylamine | 8 | 78 |
| 15 | 2,6-Difluorobenzamide | 2,6-Difluorobenzylamine | 8 | 89 |
| 16 | 4-Chlorobenzonitrile | 4-Chlorobenzylamine | 5 | 84 |