Abstract
New methods for obtaining 4-bromo-2,5-dimethoxybenzoic acid are proposed; it has been shown that the compounds to which the structures of 6-bromo-2,5-dimethoxybenzoic acid and the corresponding aldehyde were previously ascribed are actually the 4-bromo isomers.
X-Bromo-2,5-dimethoxy- benzoic acid was first obtained by the bromination of 2,4-dimethoxybenzaldebyde with subsequent oxidation1. The position of the bromine in the aromatic nucleus was not established, but it was assumed that the acid synthesized was 6-bromo-2,5-dimethoxybenzoic acid. This compound has appeared un the handbook literature under this name2. We have repeated this synthesis by Rubenstein (Scheme I) and have established that the reaction product is 4-bromo-2,5-dimethoxybenzoic acid:
Scheme I
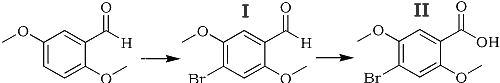
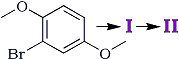
The aldehyde (I) and the acid (II) are identical with the compounds which we obtained by the formylation and oxidation of the dimethyl ether of bromohydroquinone (Scheme II).
We have also shown the structure of the acid (II) by its synthesis from the dimethyl ether of 2,5-dibromohydroquinone according to the Scheme III.
Scheme II
Scheme III
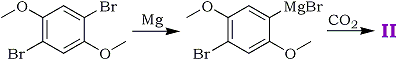
All the samples of the acid (II) melted at the same temperature, and when they were mixed in any combination, including all three acids together, the melting point did not change.
These conclusions are additionally confirmed by the PMR spectra. The PMR spectra of solutions of the various samples of the acid (II) in trifluoroacetic acid taken at a frequency of 60 MHz (Varian A-56/60 spectrometer) were completely identical. In addition to the signal of the acid proton, two singlet signals were observed of three proton units each in the positions δ 6.45 and 6.3 ppm, corresponding to the protons of the two methoxy groups, and two signals at fields of δ 2.7 and 2.8 ppm of one proton unit each with a zero spin-spin interaction corresponding to two protons of a benzene ring in the para position.
Experimental
4-Bromo-2,5-dimethoxybenzaldehyde (I)
A mixture of 70 g of bromohydroquinone dimethyl ether and 350 ml of anhydrous chlorobenzene was cooled to 0°C in a thin-walled glass vessel. Then 50 g of aluminum chloride and 14 g of anhydrous hydrocyanic acid were added. With cooling to -30 to -35°C (bath temperature), the mixture was saturated with dry hydrogen chloride until the increase in weight was 20 g, and then the vessel was tightly closed and was shaken at room temperature for 15 h. The contents of the reactor were transferred to a flask and boiled with a mixture of 80 ml of concentrated hydrochloric acid and 280 ml of water under reflux for 1 h. Then the mixture was cooled and extracted with chlorobenzene (3x100mL), the combined organic extracts were washed with 140 ml of 10% NaOH solution and then with water and were dried with MgSO4, the solvent was driven off in vacuum, and the residue was recrystallized from methanol, mp 132°C. Yield 56 g (75%).
Oxidation of the resulting aldehyde with potassium permanganate as described by Rubenstein1 yielded 4-bromo-2,5-dimethoxybenzoic acid, mp 168-169°C, yield 88%.
4-Bromo-2,5-dimethoxybenzoic Acid (II)
A solution of 20 g of the dimethyl ether of 2,5-dibromohydroquinone in 100 ml of absolute tetrahydrofuran was added dropwise to a mixture of 1.6 g of magnesium and 15 ml of hot absolute tetrahydrofuran with stirring at such a rate that the mixture boiled gently. Then it was boiled under reflux for another 1 h, after which it was cooled with water and, with stirring, 4.5-5 g of solid carbon dioxide was added to the reactor in small portions. The mixture was stirred for another 15 min, evaporated in vacuum to approximately 1/5 of its original volume, and, with cooling in ice-water, treated with an excess of hydrochloric acid; the mixture was extracted with ether, the extract was washed with water and 10% NaOH solution, and the alkaline solution was separated off and acidified with hydrochloric acid. The crystals that deposited were washed with water and recrystallized from acetic acid, mp 168-169°C. Yield 8.3 g (48%).