Benzhydrylsulphinyl-acetohydroxamic Acid (Adrafinil)1
Diphenylmethanethiol
15.2 g (0.2 mol) of thiourea and 150 ml of demineralized water are introduced into a 500 ml three-neck flask equipped with a central mechanical stirrer,
and with a dropping funnel and a condenser on the (respective) side-necks.The temperature of the reaction mixture is brought to 50°and 49.4g (0.2 mol) of
bromodiphenyl- methane are added all at once whilst continuing the heating. After refluxing for about 5 minutes, the solution, which has become limpid, is
cooled to 20°C and 200 ml of 2.5 N NaOH are then added dropwise whilst maintaining the said temperature. The temperature is then again kept at the reflex
for 30 minutes after which, when the mixture has returned to ordinary temperature (15-25°C), the aqueous solution is acidified with 45 ml of concentrated
hydrochloric acid. The supernatant oil is extracted with 250 ml of diethyl ether and the organic phase is washed with 4x80 ml of water and then dried over
magnesium sulphate. 39 g of crude diphenylmethane-thiol are thus obtained. Yield 97.5%.
Benzhydryl-thioacetic acid
10 g (0.05 mol) of diphenylmethane-thiol and 2g (0.05 mol) of NaOH dissolved in 60 ml of demineralised water are introduced successively into a 250 ml
flask equipped with a magnetic stirrer and a reflux condenser. The reactants are left in contact for 10 minutes whilst stirring, and a solution consisting
of 7g (0.075 mol) of chloroacetic acid, 3g (0.075 mol) of NaOH pellets and 60 ml of demineralized water is then added all at once. The aqueous solution is
gently warmed to about 50°C for 15 minutes, washed with 50 ml of ether, decanted and acidified with concentrated hydrochloric acid. after filtration,
10.2g of benzhydryl-thioacetic acid are thus obtained. Melting point 129-130°C. Yield 79%.
Ethyl benzhydryl-thioacetate
The following reaction mixture is heated under reflux for 7 hours: 10.2 g (0.0395 mol) of benzhydryl-thioacetic acid, 100 ml of anhydrous ethanol and
2 ml of sulphuric acid. When heating has been completed, the ethanol isevaporated in vacuo; the oily residue is taken up in 100 ml of ethyl ether and the
organic solution is then washed with water, with an aqueous sodium carbonate solution and then with water until the wash waters have a neutral pH. After
drying over sodium sulphate, the solvent is evaporated. 10.5g of ethyl benzhydryl- thioacetate are thus obtained. Yield 93%.
Benzhydryl-thioacetohydroxamic acid
The following three solutions are prepared:
- Ethyl Benzhydryl-thioacetate 10.8 g (0.0378 mol) in 40 ml methanol
- Hydroxylamine hydrochloride 5.25 g (0.0756 mol) in 40 ml methanol
- Potassium Hydroxide pellets 7.3 g (0.0134 mol) in 40 ml methanol
The solutions are heated, if necessary, until they become limpid, and when the temperatures have again fallen to below 40°C, the solution of potassium
hydroxide in methanol is poured into the solution of hydroxylamine hydrochloride in alcohol. Finally, at a temperature of about 5° to 10°C, the solution
of ethyl benzhydryl- thioacetate is added in its turn. After leaving the reactants in contact for 10 minutes, the sodium chloride is filtered off the limpid
solution obtained is kept for about 15 hours at ordinary temperature. The methanol is then evaporated under reduced pressure, the residual oil is taken up
in 100 ml of water and the aqueous solution is acidified with 3 N hydrochloric acid. The hydroxamic acid which has crystallized is filtered off, washed
with water and then dried. 9.1 g of product are obtained. Yield = 87.5%. Melting point 118-120°C.
Adrafinil (CRL 40,028)
10.4g (0.038 mol) of benzhydryl-thioacetohydroxamic acid are oxidized at 40°C, over the course of 2 hours, by means of 3.8 ml (0.038 mol) of hydrogen
peroxide of 110 volumes strength (33%), in 100 ml of acetic acid.
When the oxidation has ended, the acetic acid is evaporated under reduced pressure and the residual oil is taken up in 60 ml of ethyl acetate. The product
which has crystallized is filtered off and then purified by recrystallisation from a 3:2 (by volume) mixture of ethyl acetate and isopropyl alcohol.
8g (73%) of Adrafinil, mp 159-160°C, are thus obtained. H2O Solubility <1 g/L.
Benzhydrylsulphinylacetamide (Modafinil)2
Benzhydrylthioacetyl chloride
19.5g (0.076 mol) of benzhydrylthioacetic acid in 114 ml of benzene are placed in a three-necked flask provided with a condenser and
a dropping funnel. The mixture is heated and 19 ml of thionyl chloride are added drop by drop. Once the addition is complete, the reflux is continued for
about 1 hour, cooling and filtering are carried out and the benzene and the excess thionyl chloride and then evaporated. In this way, a clear orange oil
is obtained.
Benzhydrylthioacetamide
35 ml of ammonia in 40 ml of water are introduced into a three-necked flask provided with a condenser and a dropping funnel and the benzhydrylthioacetyl
chloride dissolved in about 100 ml of methylene chloride is added drop by drop. Once the addition is complete, the organic phase is washed with a dilute
solution of soda and dried over Na2SO4, the solvent is evaporated and the residue is taken up in diisopropyl ether; in this way, the benzhydrylthioacetamide
is crystallized. 16.8 g of product (yield 86%) are obtained. M.p. 110°C.
Modafinil (CRL 40,476)
14.39 g (0.056 mol) of benzhydrylthioacetamide are placed in a balloon flask and 60 ml of acetic acid and 5.6 ml of H2O2 (about 110 volumes, 33%) are
added. The mixture is left in contact for one night at 40°C. and about 200 ml of water are then added; the CRL 40476 crystallizes. By recrystallization
from methanol, 11.2 g of benzhydrylsulphinylacetamide are obtained. Yield: 73%. M.p. 164-66°C.
Novel Synthesis of Modafinil and its sulfone analog3
Our interest in synthesis of modified neuroactive compounds has led us to consider Modafinil (1), a stimulant and anti-narcoleptic agent that is finding increasing use in a number of neurological areas. The compound was originally prepared by a rather tedious route described in a procedure patented by L. Lafon2. More recently, its preparation has been reported by Mu et al.4 We believe that this compound has many interesting properties and possible alternative uses in addition to its recognized anti-narcoleptic actions.
Fig 1.
The chemical pathway leading to modafinil

Not having been able to obtain it from the patent holder, we proceeded to explore alternate synthetic pathways and settled on a convenient synthesis, which permitted us to produce this compound along with a primary derivative, the sulfone (2) in sufficient quantities for whole-animal studies. The current, more facile method starts with benzhydryl bromide and sodium thiolacetate in aqueous acetone, which reacts directly to form diphenylmethylthioacetic acid (3), possibly by an ionic mechanism. This resultant compound can be converted to its acid chloride that, in turn, may be used to acylate ammonia. The ensuing primary amide (4) may be gently oxidized by H2O2 to form the corresponding sulfoxide (Modafinil, 1) and, under more vigorous conditions, the modafinil sulfone (2), whose anticonvulsant and biological properties have not been described extensively in the literature. Additionally, this procedure is also uniquely suitable for large-scale preparation of Modafinil and its congeners.
One aspect of our preparation of modafinil needs further mention. When diphenylmethylthioacetamide (4) is being oxidized by H2O2, care must be taken to keep the reaction mixture cool, and workup should be done in a timely manner. Allowing the reaction to go to 24 h or longer at room temperature results in the formation of the sulfone (2). The paper by Mu et al.4 does not discuss this possibility. In our hands, the procedure stated therein led to the higher melting sulfone and not the modafinil. Our NMR data for the newly prepared modafinil preparation are in consonance with the data of the patented commercial product. It should be noted that the methylene protons in modafinil are geminally coupled and appear as a pair of doublets. This is due to the fact that the adjacent sulfoxide moiety is chiral, and therefore the methylene protons adjacent to it wind up being diastereotopic with different chemical shifts and coupling. In the sulfone 2, the methylene protons appear as a singlet due to the fact that the adjacent sulfone moiety is achiral, thus making the two protons equivalent. Modafinil 1 is, however, an equal mixture of enantiomers, as in the reported patent and publication2,4.
Experimental
The new compounds were prepared according to modified
procedures published in the patent literature. Starting materials and
solvents were obtained commercially from Fluka and/or Aldrich
Chemical Corp. Thin layer chromatography
(TLC) was performed on silica gel plates. Solvent system was EtOAc:MeOH:NH4OH,
100:10:3 by volume. Melting points are uncorrected.
Diphenylmethylthioacetic Acid (3)
Benzhydryl bromide (14.78 gm, 0.059 mole) was dissolved in 75 ml of acetone in a 250-ml round-bottomed flask. To this solution was added dropwise sodium mercaptoacetate (6.59 g, 0.058 mole) in about 60 ml of H2O; the mixture was stirred under N2 for 2 h at room temperature and was thereafter warmed at about 60–70°C for 1 h. The reaction mixture was evaporated to dryness and taken up in CH2Cl2 and saturated aqueous NaHCO3. The organic extract was rejected, and the aqueous phase was treated with acid to pH 2 and chilled. Suction filtration gave the 6.9 g of the acid (3, 46%), mp 125°C. Rf 0.2. Recrystallization from MeOH/H2O gave mp 126–128°C.
Diphenylmethylthioacetamide (4)
Diphenylmethylthioacetic acid 3 (19.5 g, 0.076 mole) in 114 ml of dry benzene was taken in a 250-ml roundbottomed flask attached to a reflux condenser, under N2 gas. To this was added thionyl chloride (19.5 ml, 0.097 mole) with a dropping funnel. The mixture was stirred at room temperature with a magnetic stirrer and refluxed for 1 h. Thereafter, the mixture was evaporated under low pressure to give a yellow oil that was taken up in about 100 ml of CH2Cl2 and filtered to yield a clear orange solution. This was chilled in ice water and added slowly to an ice-cold solution of concentrated NH4OH in H2O (40:40 ml). The ensuing mixture was stirred for 1 h and shaken well in a separatory funnel. The organic layer was dried (Na2SO4) and evaporated to dryness to give 14.39 g (54%) of the amide (4), mp 108–109°C (lit4 110°C). Rf 0.8. Recrystallization from CH3OH/H2O gave mp 109–110°C.
Diphenylmethylsulfinylacetamide (Modafinil, 1)
Diphenylmethylthioacetamide 4 (3.46 g, 0.013 mole) was taken in glacial acetic acid (14 ml) with stirring; to this was added 1.34 ml of 30% H2O2 with chilling in ice water. The mixture was left in the refrigerator for 4 h and thereafter worked up by treating it with 70 ml of ice-cold water. The precipitated material was filtered under suction and washed with ice-cold water to give 1.5 g of white crystals (43%), mp 159–160°C. Rf 0.6. Recrystallization from hot MeOH gave mp 161–162°C
Diphenylmethylsulfonylacetamide (2)
Diphenylmethylthioacetamide (2.5 g, 0.009 mole) (CAS No. 118779-53-6) was dissolved in about 12 ml of glacial acetic acid and 3 ml of 30% H2O2 and set aside overnight (16 h or more). The next day, the mixture was diluted with 100 ml of H2O and set aside to cool in the refrigerator. Upon filtration and drying, 2.1 g (80%) of 2 was obtained as a white powder. Rf 0.89. The melting point of sample after recrystallization from absolute EtOH was 195–197°C.
High-yield Synthesis of Modafinil from Benzhydrol5
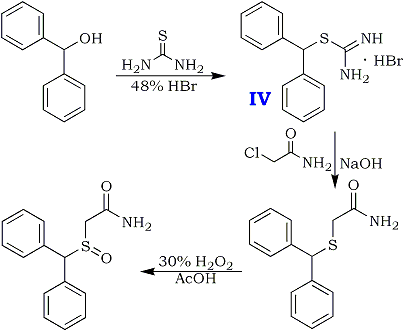
A recent patent5 describes a very easy two-step route to the Modafinil precursor diphenylmethanethioacetamide from benzhydrol (diphenylmethanol) in 90% yield and with 95% purity. A 200g batch is made in a 2000 mL vessel using water as reaction medium and ethyl acetate for recrystallization of the product.
Diphenylmethylbromide is prepared in situ from benzhydrol and react it with thiourea in a one-pot reaction to form the corresponding isothiouronium salt. The crude salt is then reacted with chloroacetamide (by generating the thiolate cation in situ), and after filtration and washing, diphenylmethylthioacetamide is isolated in excellent yield and good purity. After oxidation of the thioacetamide with hydrogen peroxide, followed by recrystallization, the overall yield of Modafinil is 67% from the benzhydrol.
(Chimimanie's Voice:) The synthesis works just as great without the nitrogen inert atmosphere (most patents do not use it at all), step two is only a hydrolysis of the thiouronium salt to the thiolate. You just have to put the salt, NaOH and heat till you got a homogenous solution, with no more solid material floating around. The following chloroacetamide SN2 reaction is a breeze too. Sometime a blue solution can bee obtained, it is nothing to worry about. In the final step, you just have to filter off the solid which did not dissolve when the crude thioacetamide is put in the GAA/H2O2, bee4 crashing the soluble one with water.
Do not forget to slurry the modafinil in EtOAc and then recrystallize it from aqueous MeOH, as the crystalline shape of modafinil is important for the kinetic and quality of effects, at least according to the patents EP0966962 and US2002043207.
Experimental
Preparation of isothiouronium Salt (IV)
Diphenylmethanol (130 g, 0.7 mole) and thiourea (65 g, 0.85 mole) are added in 0.5 L reactor charging with water (325 ml). The mixture is heated to 95°C. (an emulsion is obtained) and 48% HBr (260 gr. 3.22 mole, 4.6 equivalents) is then added gradually during 0.5 hour. The mixture is heated under reflux (106-107°C) for 0.5 hour and cooled to 80-85°C. At this temperature, the mixture is seeded with several crystals of the product and the mixture is stirred at that temperature for 0.5 hour and then cooled to 25°C. The colorless crystals are collected by filtration, washed with water (200 ml) yielding about 240 gr. of wet crude isothiouronium salt.
(Antoncho's Voice:) Assholium successfully made Modafinil by this method, but there turned out to be a mistake in the original patent text - In the preparation of IV, the quantity of HBr stated here is excessive and leads to complete hydrolysis of the initially formed isothiouronium salt. The acid should bee added until the reaction mixture turns completely clear (about half as much as the patent says) - a sort of titration. Further addition will result in precipitation of heavy stinky oil, benzhydrylmethanethiol.
Preparation of diphenylmethylthioacetamide
A 2 L reactor was charged with diphenylmethylisothiouronium bromide crude wet obtained (240 gr.) and water (700 mL) under nitrogen. The suspension was heated to 60°C and 46% aqueous NaOH solution (98 ml, 1.68 mole, 2.4 eq.) was added. The reaction mixture was heated to 85°C and stirred until all the solid was dissolved. Then, it was cooled to 60°C and chloroacetamide (80 g, 0.84 mole, 1.2 eq.) was added in five portions hour at 60-70°C during one hour. The suspension is stirred at 70°C for 4-5 hours. The mixture was filtered while warm and the cake was washed with hot water (250 ml). Diphenylmethylthioacetamide crude wet is obtained [220 gr., HPLC assay: 78%, HPLC purity: 95%, yield: 95% from diphenylmethanol]. 20g of the product was recrystallized twice from ethyl acetate, dried in vacuo to give 15g of pure title compound.
Preparation of Modafinil
A 1.0 L reactor was charged with diphenylmethylthioacetamide crude wet (220 gr.) obtained above and glacial acetic acid (610 mL). The mixture was heated to 40°C and stirred until full dissolution is achieved. 5.8% H2O2 solution (500g, 1.2 eq) was added dropwise during 0.5 hours at 40-45°C. The reaction mixture was stirred at 40-45°C for 4 hours. Then sodium metabisulfite (18.3g) in 610 mL water was added in order to quench the unreacted H2O2 and the suspension was stirred for 0.5 hours. Then the reaction mixture was cooled to 15°C and filtered. The cake was washed with water (610 mL) and dried on air to obtain crude wet Modafinil (205 g). Reslurry in refluxing ethyl acetate, followed by recrystallization from methanol:water (4:1) solution afforded pure Modafinil [125 g, HPLC assay: 99.9%, HPLC purity: 99.9%, yield: 67% (from diphenylmethanol)].