Although primary amines can be obtained from the corresponding alkyl halides by a variety of methods, only a few of these1 allow these compounds to be prepared directly from alcohols by a one-pot procedure. Recent approaches to this problem1,2 based on the Mitsunobu reaction, suffer from commercially inaccessible reagents and/or rather tedious work-ups. The method now reported judiciously combines several known reactions.
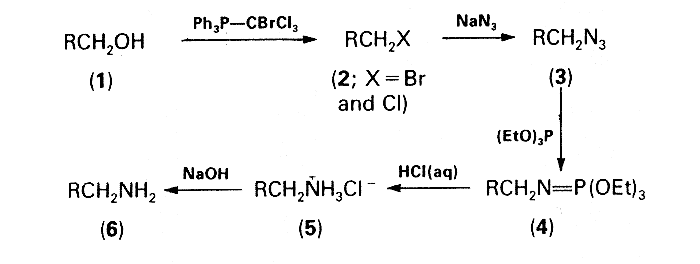
Having convincingly established the utility of transforming alkyl bromides into amines by a one-pot procedure involving the Staudinger reaction3, attention was focused upon a conversion of alcohols into bromides and/or chlorides which would then be suitable for use in this reaction without the need for isolation. Treatment of the alcohols with triphenylphosphine-tetrahalogenomethane was chosen as a reasonable approach to this problem, the reagent system triphenylphosphine-carbon tetrachloride having previously been proved very versatile for the conversion of alcohols into alkyl chlorides4. However, the relatively low reactivity of chlorides in comparison with bromides in the subsequent azidation step was discouraging. Replacement of carbon tetrachloride by bromotrichloromethane seemed a feasible method for increasing thc proportion of the more reactive bromides in the halogenated material. Moreover, in this new three-component system, Ph3P-CBrCl3 ROH, the formation of undesirable side-products, i.e. dichloromethylene- phosphorane, chloromethylene-phosphorane, and chloromethyltriphenyl phosphonium chloride, should be less than in the Appel system5 which involves only carbon tetrachloride. The combination of triphenylphosphine and bromotrichloromethane, but with different stoichiometry, has been successfully used for the preparation of dichloromethylidene-phosphoranes6. The third conceptually promising possibility of converting alcohols into the corresponding alkyl bromides by means of the two-component system carbon tetrabromide-triphenylphosphine was disqualified because of the formation of bromoform which on subsequent azidation could possibly afford highly explosive gem-diazides7 as undesirable side-products.
Table 1
Conversion of alcohols R-CH2OH (1) into
amines (6) and ammonium tosylates (7)
| Amine | R | Isolation Variant |
Amine Yield |
Tosylate Salt m.p. |
| 6a | n-C5H11 | A |
61% |
125-126°C |
| 6b | PhCH2 | A |
78% |
175-176°C |
| 6c | Ph | B |
65% |
178-180°C |
| 6d | p-MeO-Ph | B |
43% |
201-202.5°C |
| 6e | Me2CHCH2 | B |
61% |
96-97°C |
| 6f | n-C7H15 | C |
71% |
127-128°C |
| 6g | n-Bu(Et)CH | C |
67% |
62-65°C |
The effectiveness of an equimolar mixture of triphenylphosphine and bromotrichloromethane in the first step of the devised one-pot sequence depicted in the Scheme was corroborated experimentally. The mixtures of the alkyl bromides and chlorides (2) formed by the action of this reagent system on the primary alcohols (1) were then subjected to azidation and subsequent Staudinger reaction with triethyl phosphite according to the previously described procedure3. The crude iminophosphorane intermediates (4) were hydrolysed by refluxing with 20% hydrochloric acid and the solutions of the amine hydrochlorides (5) were made alkaline with sodium hydroxide. The free amines (6) were formed in good overall yields (60-70%) and characterized as their crystalline ammonium toluene-p-sulphonates. Attempts at using this procedure for converting secondary alcohols into the corresponding amines were unsuccessful: the amines were formed in very low yields (e.g. cyclohexylamine, 11%; 3-aminohexane, 21.5%) even when the azidation time was prolonged to 12 h and the amount of tetrabutylammonium bromide was increased to 10 mol%.
Experimental:
The mixture of primary alcohols (1) (0.03 mol), triphenylphosphine (8.65 g, 0.033 mol), bromotrichloromethane (6.54 g, 0.033 mol), and benzene (10 ml) was refluxed gently with stirring for 2 h. It was then cooled to room temperature and, after addition of sodium azide (3.9 g, 0.06 mol), tetrabutylammonium bromide (0.48 g, 5 mol %), and dimethylformamide (10 ml), refluxed again with stirring for 6 h. The resultant mixture was then poured into water (100 ml) and extracted with benzene (2x10 ml). The organic phase was dried (MgSO4) and treated with triethyl phosphite (5.0 g, 0.03 mol) at 25-30°C for 2 h. After this solution had been stood overnight at room temperature, 20% hydrochloric acid was added and the mixture was refluxed with stirring for 2 h.
The free amine was isolated and purified by one of the following variants.
Variant A - for low-molecular-weight, water-insoluble amines (6a), (6b)
The aqueous phase was separated, made strongly alkaline with solid sodium hydroxide, and steam-distilled. The distillate was salted out, extracted with diethyl ether (4x15 ml), and evaporated using a short Vigreux column to avoid undesirable losses of amine.
Variant B - for water-soluble amines (6c), (6d), (6e)
The aqueous phase was separated, decolorized with charcoal, made strongly alkaline with solid sodium hydroxide, and extracted with diethyl ether (5x15 ml). The extract was dried (Na2SO4) and evaporated.
Variant C - for higher-molecular-weight, water-insoluble amines (6t), (6g)
Benzene was evaporated from the hydrolysate and the volatile impurities were steam-distilled from the acidic solution. The solution was made strongly alkaline with solid sodium hydroxide and steam-distilled again. The distillate was extracted with diethyl ether (4x15 ml), dried (Na2SO4), and evaporated.
The free amines (6) were treated with equimolar amounts of para-toluenesulphonic acid in ethanol. Crystalline ammonium para-toluenesulphonates (7) were precipitated with diethyl ether and purified by dissolving in a small amount of ethanol and then reprecipitating with an excess of diethyl ether. The yields of the free amines (6) and the mp's and analytical data of the ammonium para-toluenesulphonates (7), are compiled in the Table.