Abstract
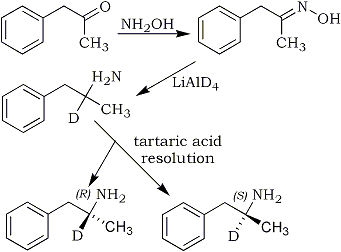
In order to investigate the isotope effect on the enzymatic deamination of amphetamine, deuterio-l-amphetamine, d1 sulfate was synthesized by the selective reduction of phenyl-2-propanone oxime with lithium aluminum deuteride, followed by resolution of the racemic product. The yield of racemic amphetamine was 46% when approximately equimolar quantities of the oxime and LiAlD4 were employed. The identity of the deuterated amphetamine base was confirmed by determination of the physical properties of the racemic mixture. From NMR data, deuterium was found exclusively in the methine hydrogen position at a purity of greater than 99 atom%. The optical isomers of deuterioamphetamine were resolved through repeated fractional crystallization of the d-amphetamine-d-bitartrate and l-amphetamine-l-bitartrate diastereomers. The isomers were found to possess 97-99% optical purity, based on values for pure isomers of protioamphetamine. In vitro metabolism studies indicate that a significant deuterium isotope effect operates in the oxidative deamination of deuterio-l-amphetamine. Under specified conditions, the ratios of apparent rate constants (kH/kD) based on initial velocities, yield a value of 2.0±0.3.
The deuterium-isotope effect has been applied with significant success to the study of experimental pharmacology. Belleau, in a recent monograph1, has reviewed the applications of the isotope effect and has proposed a mechanism for certain bioreceptor-substrate interactions which has importance in the studies of drug response and metabolism. The concept of a pseudo-primary isotope effect has been developed to explain the large isotope effect observed in some systems which involve a Michaelis complex formation. The existence of a deuterium-isotope effect in the metabolism of various therapeutic agents has led these investigators to consider the possibility that such an effect exists in the oxidative deamination of amphetamine.
Although sustained retention of a drug does not necessarily mean an increased duration of action, the correlation has frequently been observed. Beckett et al.2 have reported an increased duration of wakefulness in human volunteers receiving d-amphetamine, which could be correlated with a depressed excretion rate of administered drug. While various paths of excretion and tissue storage are prime considerations in the fate of a drug, the existence and extent of metabolic activity toward such a drug can be a major factor in the total elimination process. Quinn et al.3 have demonstrated that a correlation exists between depressed microsomal enzymatic activity and extended hexobarbital sleeping time.
A similar microsomal preparation has been found to effect the N-demethylation of morphine. Ellison et al.7 studied the pharmacological properties and kinetics of N-demethylation of morphine which has been substituted with deuterium in the N-methyl position. Deuterium substitution effected a slower rate of demethylation and a significant reduction in pharmacologic potency. Belleau1 explained that N-demethylation must occur at the receptor site as a requisite to the biological response to morphine. Should this reaction be subject to an isotope effect, the respective response would be similarly affected.
Substitution of deuterium into compounds by reduction has been facilitated by the availability of lithium aluminum deuteride. The reduction of the appropriate precursor with lithium aluminum deuteride eliminates the more tedious aspects of catalytic hydrogenation (D2 gas) procedures. Although several different chemical syntheses involving various precursors, might result in substitution of deuterium in the α-position of amphetamine, the reduction of phenyl-2-propanone oxime reported by Larsson8 was chosen for this study. Resolution of the product, racemic deuterio-amphetamine, was effected using the method of Blackburn and Burghard9. The cyclic crystallization of d-amphetamine-d-bitartrate and l-amphetamine-l-bitartrate diastereomers is a convenient procedure for obtaining maximum optical purity and recovery.
Scheme I
Synthesis of deuterio-l-amphetamine, d1 sulfate.
It is apparent from a consideration of the substrate structure and that of the proposed metabolite, benzyl methyl ketone, that the C—D bond of α-deuterioamphetamine (see Scheme I) will be broken sometime during an enzymatic oxidative deamination. If this event is involved in the rate-limiting process, a significant deuterium isotope effect might well be anticipated. The relevance of such an effect to amphetamine therapy is interesting. The normally low excretion rate of amphetamine is significantly depressed under conditions of high urinary pH2. Under these conditions, the duration of certain pharmacological responses to this drug could be determined by the rate at which it is metabolized.
The purpose of the present investigation was to synthesize deuterio-l-amphetamine, d1 sulfate which possesses maximum optical and isotopic purity, and to determine the existence of a deuterium-isotope effect in the oxidative deamination of this drug.
Experimental
Phenyl-2-propanone Oxime
The microsynthetic procedure of Wilson10 was modified to prepare phenyl-2-propanone oxime by the addition-dehydration reaction between phenyl-2-propanone and hydroxylamine (Scheme I). The product of the reaction, phenyl-2-propanone oxime, was extracted into ether, washed twice with saturated calcium chloride solution, and dried over anhydrous magnesium sulfate. The solvent was removed at reduced pressure using a rotary evaporator, and the residue was distilled, giving a colorless viscous liquid boiling at 81–84°C/0.15mmHg [reported11 bp 99°C/2mmHg]. The crude oxime was recrystallized from n-hexane to a constant melting point, 69.0°C [reported12 mp 70.0°C]. The yield of recrystallized oxime was 85%.
Deuterio-dl-amphetamine, d1
The hydrochloride derivative of deuterio-dl-amphetamine was prepared by the dropwise addition of 2 mL of HCl-saturated absolute ethanol to a cooled flask (5°C) containing 0.30 g of deuterio-dl-amphetamine in 75 ml. of anhydrous ether. The crystals were filtered with suction, washed several times with dry ether, and dried over P2O5. The melting points of deuterio-dl-amphetamine HCl and an authentic protio-dl-amphetamine HCl sample were compared: deuterio-dl-amphetamine HCl mp 146–148°C; protio-dl-amphetamine HCl, mp 146–148°C [reported15 mp 145-147°C].
Resolution of Deuterio-dl-amphetamine
All determinations of optical purity were based on the observed specific rotations of authentic samples of the appropriate protio-amphetamine-bitartrate.
Resolution of 10.5 g. (77 mmoles) of racemic deuterio-amphetamine, accumulated during three separate runs, was achieved by the cyclic crystallization of the bitartrate diastereomers. A portion of the racemic deuterio-amphetamine (5.56 g.; 41 mmoles) was dissolved in 70 ml of 91% isopropanol. The solution was heated to 80°C, 6.38 g. (42.5 mmoles) of l-tartaric acid was added, and the temperature was maintained at 80° until the acid dissolved. The solution was allowed to cool slowly (ca. 1 hr.) to 60°C and maintained at that temperature for 24 hr., whereupon crystals formed. After decantation of the mother liquor, the crystalline l-deuterio-amphetamine-l-bitartrate was filtered and washed with 13 mL of 91% isopropanol at 60°C. Optical purity of the crystalline deuterio-l-amphetamine-l-bitartrate (3.84 g.) was approximately 90%. After one recrystallization at 60° from 91% isopropanol, 2.14 g. of the diastereomer was obtained with an optical purity of 98% (equivalent to 37% of the l-isomer present in 5.56 g. of racemic deuterioamphetamine). The mother liquor, which was rich in d-deuterioamphetamine, and all rinses were combined and the solvent was removed in vacuo. The salt was dissolved in 15 ml. of water, made strongly alkaline with 25% sodium hydroxide, and extracted with 300 ml. of ether. Approximately 5.0 g. of d-tartaric acid was dissolved in a 91% isopropanol solution of the d-rich deuterio-amphetamine base which had been recovered from the d-rich deuterio-amphetamine-l-bitartrate salt. Crystallization of d-amphetamine-d-bitartrate was effected at 60°C in a manner identical to that used for the l-bitartrate diastereomer. A yield of 3.1 g. of deuterio-d-amphetamine-d-bitartrate was obtained with an optical purity of 98%. This crystallization cycle was repeated three times, adding the remainder of racemic deuterio-amphetamine (4.94g) after the third crystallization. The combined deuterio-l-amphetamine-l-bitartrate fractions yielded 4.94g of the diastereomer, [α]25D -29.5° (2%, H2O), equivalent to 45% of the l-isomer present in 10.5g of racemic deuterio-amphetamine base. The yield of deuterio-d-amphetamine-d-bitartrate was 5.91g (54%), [α]25D +29.8° (2%, H2O). The synthesized isomers possess an optical purity of at least 98% when compared to authentic samples of each diastereomer. Specific rotation of the standard samples was -30.3° and +30.5° for the l- and d- forms, respectively.
The diastereomer salt of each isomer was dissolved in 15 mL of water, made strongly alkaline with 25% sodium hydroxide, and extracted with 300 ml. of ether. The solvent was removed in vacuo. The sulfate salt of each isomer was prepared by the addition of an equivalent amount of 50% sulfuric acid to a solution of the respective deuterio-amphetamine base in 91% isopropanol. The crystals of each isomer were filtered with suction and dried over P2O5. A recrystallized sample of deuterio-l-amphetamine d1 sulfate was submitted for elemental analysis.
Results and Discussion
Table I
LAH:Oxime Ratio Yield Effect
LAH/Oxime |
Yield |
Amph/LAH |
2.20 : 1 |
75.0% |
0.34 |
1.65 : 1 |
67.0% |
0.41 |
1.10 : 1 |
48.5% |
0.44 |
Preliminary experiments using lithium aluminum hydride indicated that the most favorable ratio of reducing agent to oxime was 1.65:1, based on the oxime precursor (see Table I). However, the most favorable ratio based on the reducing agent was determined to be 1.1:1. This ratio was chosen for the preparation of deuterio-dl-amphetamine. No significant difference in yield of amphetamine was observed when lithium aluminum deuteride was substituted for lithium aluminum hydride in the preparation of the product.
Identity of the product as deuterio-amphetamine was confirmed by the determination of several physical constants. These determinations were found to be in close agreement with literature values for protio-amphetamine. As expected, the values for deuterio-amphetamine (mol. wt. 136.21) are not greatly different from the constants for authentic protio-amphetamine (mol. wt. 135.21). Resolution of deuterio-dl-amphetamine yielded isomers which exhibited optical activity similar to that of the protio-amphetamine isomers.
Table II
Summary of Chemical Shifts (δ ppm from TMS)

A most important aspect of this study concerned the positional isotope analysis of deuterio-dl-amphetamine. The NMR spectra of deuterio-dl-amphetamine were the basis for this analysis. Table II contains a summary of the chemical shifts for deuterio-dl-amphetamine and protio-dl-amphetamine. As expected, the methyl signal of deuterio-dl-amphetamine is reduced to a singlet and the methine signal of protio-dl-amphetamine is absent from a single scan of deuterio-dl-amphetamine. Spectra of a (neat) sample of deuterio-dl-amphetamine in the region of 2.5-3.0 p.p.m. (methine), obtained at high-spectrum amplitude, confirm the replacement of the proton by deuterium at this molecular position. The methine signal was totally absent, in spectra obtained for deuterio-dl-amphetamine. The identity of deuterio-dl-amphetamine, represented in Scheme I, has been assumed to be established. Isotopic purity at the α-position appears to be greater than 99%.
An apparent solvent effect on the methylene signal of deuterio-dl-amphetamine was observed, illustrating the nonequivalence of the methylene protons. The methylene signal of deuterio-dl-amphetamine appears as a singlet in a neat sample, a partially split broad peak in carbon tetrachloride, and a doublet in deuterochloroform. Similar solvent effects have been observed with protio-amphetamine18.
Phenyl-2-propanone oxime is most commonly recovered as a viscous oil, but crystallizes on standing. The melting point reported for this compound is 70°C12. Spin-splitting analysis of phenyl-2-propanone oxime supports evidence, first reported by Lustig19, for the simultaneous existence of syn- and anti-isomers in the oil. Lustig suggests that, "nonlinearity of the C=NO— group gives rise to this isomerism", which locates the methylene protons of the isomers in a nonequivalent magnetic environment. The nonequivalent methylene protons are represented by single peaks at 3.50 p.p.m. (1.5 H) and 3.75 p.p.m. (0.5 H). Integration of the signals at 3.50 and 3.75 p.p.m. indicates that the oil consists of a nonequilibrium mixture of two isomers (syn and anti), present in a ratio of approximately three to one. Lustig further suggests that the crystalline phenyl-2-propanone oxime (mp 70°C) gives rise to the stronger component (3.50 p.p.m.) of the methylene signal. Isolation of the second isomer has not been reported.