Acetyl Chloride, Acetic anhydride and Propionic AnhydrideAcetyl Chloride
Connect a dry 250 ml three neck flask with a distillation adapter, condenser and addition funnel. Use a 250 ml rb flask for the receiver connected to the condenser with a vacuum adapter. Use the vacuum adapter to make a gas trap with rubber tubing to an anhydrous CaCl2 drying tube exiting to an inverted funnel suspended over a beaker of water. This important stage is to protect the distillate from water while absorbing HCl evolved in the reaction. Place 54 g. (52 ml.) of glacial acetic acid in the three neck flask, close with a stopper and add slowly through the addition funnel 46 g. (29 ml.) of phosphorus trichloride. Cool the flask by immersion in a water bath. Mix the reactants thorougly and, after allowing the mixture to stand for ten minutes, heat the water bath to 40-50°C and maintain it at this temperature for thirty minutes, with occasional swirling of the flask. During the heating the liquid usually separates into two layers. Acetyl chloride forms the upper layer. Distill the acetyl chloride by heating the water bath to boiling and maintaining it at that temperature as long as any liquid passes over. Cool the receiver in an ice bath during the distillation. The syrupy residue in the distilling flask is phosphorous acid, which is discarded. CAUTION: The reaction mixture must not be overheated since this will lead to formation of phosphine, which is spontaneously flammable in contact with air. To the distillate add two drops of glacial acetic acid, to destroy mixed phosphorous-acetic anhydrides that would cause turbidity to develop on standing. Redistill the acetyl chloride from a distillation apparatus arranged as before except without the addition funnel and with a thermometer. Collect separately in a dry receiver the portion boiling at 50-56°C and transfer it to a dry weighed glass-stoppered bottle. Acetyl chloride attacks corks and rubber stoppers. The yield is 44-56g. Acetic Anhydride
The apparatus for this process is similar to that used for the preparation of acetyl chloride. The same precautions for exclusion of moisture must be observed but it is unnecessary to provide the inverted funnel arrangement, since hydrogen chloride is not evolved. In a 250ml. rb flask fitted as for making acetyl chloride, place 60 g. of finely pulverized anhydrous sodium acetate (*) Arrange in place the condenser, dry receiving flask and drying tube; the receiver need not be cooled. Check to insure that all connections are tight. Cool the reaction flask in a bath of cold water and add dropwise, through the addition funnel, 40 g. (36 ml.) of acetyl chloride. After the addition has been completed, remove the water bath and shake the flask to obtain good mixing of the reactants. Recheck the connections for tightness. Dry the outside of the flask with a towel and heat it with a mantle or oil bath. Continue the heating until no more distillate comes over but do not overheat the solid residue. To the distillate add 4-6 g. of finely powdered anhydrous sodium acetate, to react with a small amount of acetyl chloride that may be present. Add a boiling chip and redistill the crude acetic anhydride. Collect the product distilling at 132-138? in a dry flask. The yield is 30-40 g. * Note: Commercial anhydrous sodium acetate usually contains some moisture. To remove moisture fuse 70-80 g. in a casserole (either in an ordinary oven, or a microwave) and stir until no more water is evolved. Do not overheat! Cool and quickly pulverize. Store in a tightly sealed container before use. Acetic Anhydride and Propionic AnhydrideDrone #342: Here's what everyone's looking for. Some things are a little wierd about it, like the fact in the acetic anhydride synth they use a small quantity of acetic anhydride as a solvent. However, as one sees in propionic anhydride, such a solvent may not be necessary. From "Chemistry & Industry", 1945, p. 382; "LABORATORY METHOD FOR THE PREPARATION OF ORGANIC ACID ANHYDRIDES" by Jehuda Orshansky and Eliahu Bograchov. "...(1) Acetic anhydride. To 50 g. acetic anhydride in a round-bottomed flask of 1500 cc. capacity, placed in cold water, 440 g. of powdered sodium acetate (dried by fusion at 320 C) and at the same time a solution of 22. g of sulfur in 320 g. bromine is added while stirring. The operation takes about 30 minutes. "The mixture is then stirred for a further 5 minutes, after which period the initially dark brownish-red colour has changed into pale yellow, and the anhydride is distilled off from a water bath under reduced pressure. The crude anhydrie (310 g) is redistilled under normal pressure, and the fraction boiling between 134-138 C is collected. Yield, 295 g. of 98% purity = 87.5%. The so purified anhydride contains neither bromine nor sulphur compounds and leaves no residue on evaporation..." "(2) Propionic anhydride. To 40 g. fused and powdered sodium propionate in a flask of 250 cc. capacity a solution of 2 g. sulphur in 22 g. bromine was added while stirring. The temperature was kept at about 50 C. When the operation was completed, the anhydride was distilled off in vacuo. The crude product (25g.) was fractioned under normal pressure, and the fraction 155-156 C was collected. Yield, 23 g. propionic anhdride of 90% purity = 85%..." The paper mentions that other alkali metals and alkali earth metals work just fine. Calcium propionate is a food preservative added to cheap white bread to keep it from molding. With these nuggets of information, the most closely watched reagent on the DEA's watched list, propionic anhydride, just turned OTC. I can almost see the fentanyl analogs clogging the opioid market already. The one reason, and justafiably so, they poo-poo using chlorine (which does indeed work nicely) is that its a hassle to work with, especially considering the fact that they'd be adding a gas to a solid to make a liquid. I propose that perhaps with the use of chloroform as a solvent, chlorine could be bubbled in readily, and the reaction would go as previously stated. I assume they tried to avoid using extra solvents in hopes of staying away from azeotropes messing up their products' purity, so this may or may not work, depending on what you're trying to do. Iudexk: To 66.5g fused AcONa (made from AcOH + NaOH) was added a soln. of 3.3g S in 48.4g Br2 over 5mins, under manual stirring. Brown colour disappears rapidly on stirring to give slightly off-white mixture. Stirring continued for another 15mins. Mixture has v. strange consistancy; becomes almost liquid while stirring but as soon as one stops stirring it becomes solid again. Thus formed sludge was scooped up and dumped in a distillation flask (spilling much in the process - do this rxn in the same flask you're gonna distil from), and distilled. Yield of Ac2O = 29.2g as a clear pungent liquid. Acetic anhydride from sodium acetate, using S2Cl2Translated from russian by Antoncho 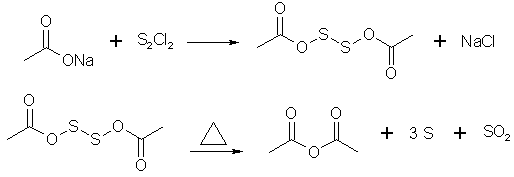
Prepare 100g freshly fused NaOAc and 65g S2Cl2. A small quantity of NaOAc is placed in a thin-walled glass cooled in an ice bath. To this is added some sulfur chloride, the mixtr is vigorously stirred w/a wooden spatula, not allowing the temp to rise. Then some NaOAc is added again, and the process is repeated several times until all is mixed in. The semi-liquid mass is transferred into a 1 liter RBF. The previous operation is repeated 4 times, so that 400g NaOAc and 260 g S2Cl2 total are taken into work. The RBF is then equipped w/a reflux condenser and gently heated on a water bath to ~80-85°C. As soon as the rxn starts, the heating is removed, and in case the rxn gets too vigorous it's cooled w/cold water. After 20-30 min SO2 evolution ceases and the mixture is heated for 10 more min's on a boiling water bath. The rxn product is then distilled off under vacuum, then fractionally re-distilled at ordinary pressure, collecting the fraction boiling between 132-142°C. For further purification it's distilled with 2-3% KMnO4 or K2Cr2O7 for breakage of sulfurous contaminants (test for their presence: 1 ml of the distillate upon neutralization w/pure NH3 mustn't give a dark precipitate on treatment with Pb(AcO)2) The yield is ~90% based on S2Cl2. Another similar synthesis, generating sulfur chloride in situ: Mix quickly and thoroughly 205-215 g of pulverized fine NaOAc and 10g dry sulfur powder, the mixture is quickly transferred to a wide-mouth 1 liter RBF and wetted w/25 ml acetic anhydride. Into the flask through the rubber cork extend 1) a wide tube for chlorine in-flow 2) an overhead stirrer, which is sealed with the aid of a piece of rubber tubing greased with vaseline and 3) an out-leading tube for excess Cl2 release. The flask is immersed in an ice-bath. Chlorine is initially passed in very carefully, with frequent stirring or shaking, over the passage of time the rxn gets hotter and more and more liquid, so the stirrer may bee after some time rotated with a motor. Chlorine stream should bee regulated so that almost all of it is absorbed. When the reaction mixture stops heating and Cl2 is no longer taken up, the rxn contents are distilled in vacuo at oil bath temp ~150-180°C, then redistilled at ordinary pressure, collecting the fraction boiling between 132 and 142°C. Yield ~90%. A Mild Method for the Synthesis of Carboxylic AnhydridesGeneral Procedure, exemplified for Propionic Anhydride:Propionic Acid + Methanesulfonyl chloride --Et3N--> Propionic Anhydride To a stirred solution of propionic acid (2 mmol) and methanesulphonyl chloride (1.05 mmol) in dry THF (10 ml) at -15°C under nitrogen was added triethylamine (3.5 mmol) in THF (5 ml) slowly over 20 minutes. After stirring 1 hour at -15°C the reaction mixture was allowed to warm to room temperature. The reaction mixture was filtered through Celite and the solvent was evaporated off under reduced pressure. The anhydride was isolated by short-path distillation (yield: 85 %, bp: 70-72°C / 20 Torr (lit. bp: 67.5°C / 18 Torr)). Reference:
This method is fairly general. The authors prepared several anhydrides, in good to excellent yield. There are two methods of isolation (used depending on the anhydride made). Method of isolation AThe solvent was evaporated under reduced pressure and the residue was partitioned between ether or ethyl acetate (50 ml) and a cold saturated solution of sodium hydrogen carbonate (50 ml). The organic phase was washed successively with saturated sodium hydrogen carbonate solution and brine, dried (MgSO[sub]4[/sub]), and concentrated in vacuo to afford the anhydride. Method of isolation B(implemented in the propionic anhydride synthesis) The reaction mixture was filtered through Celite and the solvent was evaporated off under reduced pressure. The anhydride was isolated by short-path distillation. Synthesis of internal anhydridesTo a stirred solution of phthalic acid (0.332 g, 2 mmol) in dry THF (12 ml) was added triethylamine (0.202 g, 2 mmol) under nitrogen. The reaction mixture was cooled to - 20 °C. After stirring for 15 minutes, methanesulphonyl chloride was added (0.24 g, 2.1 mmol) followed by triethylamine (0.404 g, 4 mmol) in THF (5 ml) over 15 minutes. The reaction mixture was stirred at -15°C for 2 hours. The product was isolated using method A (0.275 g,, 93 %, m.p. 130-131°C) Other ExamplesAcid: MeCH=CHCOOH Yield anhydride: 87% Method of isolation: B Acid: PriCOOH Yield anhydride: 89% Method of isolation: A Acid: Me(CH2)7CH=CHCOOH Yield anhydride: 95% Method of isolation: A Acid: PhCOOH Yield anhydride: 92% Method of isolation: A Acid: 4-O2N-C6H4COOH Yield anhydride: 82% Method of isolation: B Acid: PhCH=CHCOOH Yield anhydride: 85% Method of isolation: B Acid: 4-MeO-C6H4COOH Yield anhydride: 94% Method of isolation: B Acid: 2-Cl-C6H4COOH Yield anhydride: 93% Method of isolation: A |