Abstract
The preparation of the optical antipodes of α-aminopropiophenone (cathinone) from norephedrine and an improved large-scale resolution of norephedrine are described. The characterization of cathinone and its salts is discussed.
The chewing of the leaves of Catha edulis Forsk (Khat) by the natives of several Asian and African countries to provide rapid stimulation is extremely prevalent1 and has been considered to be a serious problem of drug dependence not unlike that associated with amphetamine2. In fact, on the basis of the observations of Eddy et al.3 the United Nations Narcotics Laboratory undertook research on the chemistry of Khat and its components2.
Earlier in this century (+)-norpseudoephedrine, a CNS-active compound, was identified among the basic alkaloid components of Khat4. Later investigations5-8 revealed the presence of other bases, but the isolation and characterization of the main constituent has only been reported within the last few years by the United Nations Narcotics Laboratory9,10. Specifically, the presence of (-)-α-aminopropiophenone9 and its absolute configuration10, as well as the presence of 3,6-dimethyl-2,5-diphenylpyrazine9 were reported. Since this information is only available in the United Nations Documents which are somewhat hard to come by11, it will be briefly summarized here. Extraction of freeze-dried plant material with methanol, followed by separation of the nonpolar and weakly basic species and purification by acid-base extractions gave an odiferous yellow oil which, after treatment with oxalic acid, gave a solid: mp 157-160°C9. This compound was identified as α-amino- propiophenone oxalate by its UV, IR, 1H-NMR, and mass spectra9. In another reports the absolute configuration of the plant extract was deduced from comparison of the CD curve observed for a separately isolated sample of oxalate salt with those of model compounds. It was concluded10 that natural α-aminopropiophenone has the S configuration, which is biogenetically consistent with the configurations of both (+)-norpseudo- ephedrine and (-)-norephedrine at the corresponding asymmetric center.
These reports do, however, contain some puzzling results. The stereospecific synthesis of (-)-α-aminopropiophenone by bromination of propiophenone followed by the Gabriel synthesis is claimed10. Indeed, the melting point (175°C) reported for the hydrochloride salt of the synthetic product differs substantially from that reported previously for racemic α-amino- propiophenone hydrochloride (184°C (12), and 187°C13 and is in agreement with the melting point reported by Takamatsu (175-176°C14) for (+)-α- aminopropiophenone hydrochloride. Furthermore, it was reportedly that when this salt was converted to an amine oxalate, a solid with a melting point of 172-175°C, which showed no melting point depression with natural cathinone oxalate, was obtained.
These results seem to suggest that the stereospecific synthesis of (-)-α-aminopropiophenone hydrochloride from propiophenone had been accomplished without the benefit of optically active reagents, solvents, or resolution. Other apparent inconsistencies appear in the literature relating to α-aminopropiophenone. For example, the report14 that treatment of α-aminopropiophenone with ethanolic hydrochloric acid gave (+)-α-amino-propiophenone hydrochloride, mp 175-176°C, would appear to be an error presumably the l-mandelate salt of (+)-α-aminopropiophenone was treated with ethanolic hydrochloric acid.
In the light of increased interest in expanding the pharmacological and medical investigations of the major constituents of Khat2 and because known methods of resolution of racemic α-aminopropiophenone were inadequate for the preparation of gram quantities of the required antipodes of α-aminopropiophenone, we undertook the development of an improved synthetic pathway to optically active α-aminopropiophenone. At the same time, having pure α-aminopropiophenone and its optical antipodes in hand, we were able to establish some of the properties and chemical behavior of α-aminopropiophenone and of its salts and to explain some of the literature discrepancies.
Results and Discussion
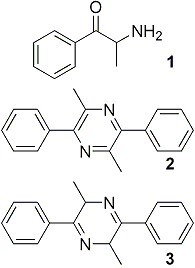
Attempts to resolve (-)-α-aminopropiophenone via the mandelate salt by following the reported procedures7,14 yielded only minute quantities of product in spite of the care taken to exclude light. Somewhat better results were obtained with tartaric acid. Thus, (+)-α-aminopropiophenone crystallized with (-)-tartaric acid and the (-) enantiomer crystallized with (+)-tartaric acid. For both antipodes the yields were small, probably due to the strong tendency of α-aminopropiophenone (1) to cyclize to 3,6-dimethyl-2,5-diphenyldihydropyrazine (3), with subsequent oxidation to 3,6-dimethyl-2,5-diphenylpyrazine (2) when it is not stabilized by the presence of strong acids13.
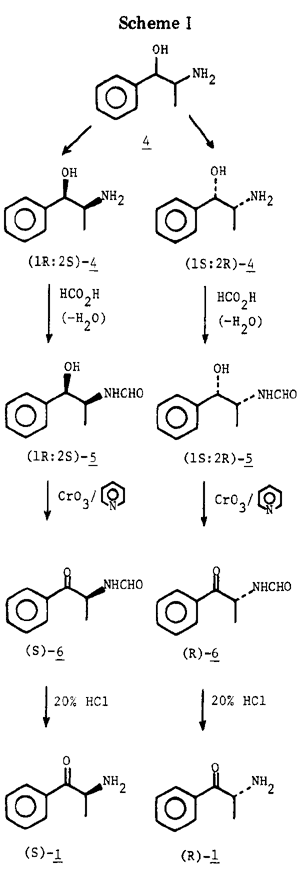
Consequently, an alternative route to optically active cathinone was developed. Norephedrine was resolved in high yield into its (+) and (-) antipodes with O,O-dibenzoyl- d-tartaric acid15. Each enantiomer was converted to its N-formyl derivative and oxidized with chromium trioxide in pyridine. Hydrolysis with 20% hydrochloric acid at 40°C gave optically pure α- aminopropiophenone hydrochloride without racemization. In this manner there was obtained, from racemic norephedrine, (-)-α-aminopropiophenone in 39% overall yield and the (+) enantiomer in 40% overall yield. It should be noted that use of an N-acetyl blocking group in the oxidation of norephedrine to α-aminopropiophenone was unsuitable because removal of the acetyl group led to racemization (~60%). Similarly, if deprotection of N-formyl-α-aminopropiophenone was carried out with weaker acid (<20% HCl) and at higher temperature (>40°C), racemization was observed.
An approach to the stereospecific synthesis of (S)-cathinone from (S)-N-(methoxycarbonyl)alanine has been recently published17. The Friedel-Crafts yield was reported to be 50-60%; assuming a 90% yield in removal of the N-methoxycarbonyl protecting group, the overall yield obtained would be slightly higher than that obtained by our method. However, whereas our approach leads to both enantiomers on using a natural (and therefore relatively inexpensive) tartaric acid derivative, preparation of (R)- cathinone by the procedure of McClure et al17 requires nonnatural alanine.
Surprisingly, the melting points obtained by us for racemic and optically active α-aminopropiophenone hydrochloride were practically the same (racemic, mp 190-191°C; optically active, mp 189-191°C). Furthermore, our melting points for the optical antipodes were substantially higher than those previously reported (175-176°C)13. Our experiments suggest that the low melting point was observed when the samples were insufficiently dried.
Because it had been found that optically active α-aminopropiophenone racemized readily in the absence of strong acids18, it occurred to us that the plant extract obtained by using dilute acetic acid at room temperature might have racemized to some extent during the extraction procedure and/or while present as the salt of a weak acid such as oxalic acid. The hydrochloride salts of (-)-α-aminopropiophenone and of the racemate were therefore converted to the oxalate salts. The optically active and racemic oxalate salts had very similar melting points (optically active, mp 173-175°C; racemic, mp 172-173°C), as we had found for the hydrochloride salts. In addition, they did not exhibit melting point depression when admixed. Thus, although optical resolution by direct crystallization of mixtures of interconverting enantiomers is possible in principle19, the strong tendency of α-aminopropiophenone to racemize and/or cyclize when it is not stabilized by the presence of a strong acid13,18 may account for the apparent identity of the oxalate salt of the plant extract and of synthetic α-aminopropiophenone observed10. Clearly, in this case the melting points cannot serve as criteria for optical purity.
Conclusions
- The optical antipodes of α-aminopropiophenone can be prepared in high yield by oxidation of optically active N-formylnorephedrine, followed by deblocking of the amino group. The resolution of nor-ephedrine is best carried out by using O,O-dibenzoyltartaric acid.
- Melting points cannot be used as criteria of the optical purity of cathinone derivatives because of the (a) similarity of the melting points of the racemic and optically active compound and (b) lack of melting point depression upon admixing of opposite antipodes and/or racemic and optically active compounds.
- The cathinone free base racemizes and dimerizes readily in a hydroxylic medium. Similar behavior is observed for solutions of the oxalate salt but at a somewhat reduced rate; therefore, the plant extract of Schorno10 may have been partially racemized.
- The cathinone free base is fairly stable as a dilute solution in nonpolar, nonhydroxylic media, and, consequently, strong acid salts of optically active cathinone can be interconverted without racemization.
Experimental
Resolution of Norephedrine
A solution of 80 g (0.53 mol) of (-)-norephedrine in 400 mL of EtOH was combined at 60°C with a solution of 95 g (0.27 mol) of O,O-dibenzoyl-d-tartaric acid in a mixture of 100 mL of EtOH and 250 mL of i-PrOH. When the mixture had cooled slightly, some seed crystals were added, and the salt was allowed to crystallize undisturbed for 3 days. The clear supernatant was carefully decanted from the crystal cake of (+)-norephedrine O,O-dibenzoyl-d-tartrate. The solid was washed thoroughly with EtOH/i-PrOH, and the washings were filtered into the supernatant. This operation usually caused the crystallization of (-)-norephedrine O,O-dibenzoyl-d-tartrate in the filtrate. After 2 days, the (-)-norephedrine salt was collected by filtration, washed with THF and dried at 120°C. The yield was 75g (86%) of (-)-norephedrine O,O-dibenzoyl-d-tartrate and 68g (78%) of (+)-norephedrine O,O-dibenzoyl-d-tartrate. Both products had a melting point of 190-200°C (dec). For conversion to the corresponding hydrochlorides, the salts were each treated with 150 mL of 7% ethanolic hydrochloric acid at 60°C for 30 min. After evaporation of the solvent, the residue was triturated with THF which caused spontaneous crystallization. The solid was washed with THF and EtOH and vacuum dried. From the filtrates was obtained a second crop upon cooling. The following results were obtained. (-)-Norephedrine hydrochloride: 40 g (81%); mp 170-172°C; (lit.15 mp 171-172°C). (+)-Norephedrine hydrochloride: 37 g (74%); mp 170-172°C (lit.15 mp 171-172°C).
(+)-N-Formylnorephedrine
A solution of 110 g (0.73 mol) of (+)-norephedrine in 250 mL of THF was cooled in an ice bath while a mixture of 35 g (0.73 mol) of 95% formic acid was added dropwise with stirring over 1 h. The pasty product was diluted with ether to give 134 g (94%) of the crystaLine formate of (+)-norephedrine. The formate was dissolved in 700 mL of toluene and refluxed for 2 days, with the water produced being separated in a Dean-Stark trap. The syrupy product (+)-N-Formylnorephedrine, which remained after evaporation of the solvent and vacuum drying, weighed 115g (92%).
(+)-N-Formyl-alpha-amino-Propiophenone (23)
An ice-cold mixture of 3 L of CH2Cl2 and 190 g (2.4 mol) of pyridine was treated with 120 g (1.2 mol) of dry CrO3 which was added in portions within a 30-min period. The purple-brown mixture was stirred at room temperature for 2 h. A solution of 38 g (0.21 mol) of (+)-N-Formylnorephedrine in 200 mL CH2Cl2 was added to the CrO3 mixture all at once with vigorous mechanical stirring. After 15 min, the yellow solution was decanted from a black, sticky precipitate and immediately extracted with 1 L of 5% NaOH solution followed by 1 L of 10% HCl solution. The organic phase was filtered through a bed of Na2SO4 and evaporated to a clear, light yellow syrup. After vacuum drying overnight, the product (+)-N-Formyl-α-amino-propiophenone weighed 31 g (83% yield). The material was sufficiently pure for further synthesis.
(+)-alpha-Amino-propiophenone Hydrochloride
A suspension of 21 g (0.12 mol) of (+)-N-Formyl-α-Propiophenone in 200 mL of 20% HCl was vigorously stirred at 40°C until a clear solution resulted (~5 h). The mixture was evaporated to dryness, and the residue was crystallized from i-PrOH-Et2O. After the sample was dried overnight (P2O5-KOH), 16 g (72%) of (+)-α-Aminopropiophenone HCl was obtained: mp 186-189°C. One recrystallization from i-PrOH-THF gave a product: mp 186-189°C.
(-)-N-Formylnorephedrine
The free base isolated from 115 g (0.62 mol) of (-)-norephedrine hydrochloride by CHCl3-aqueous NaHCO3 partition was dissolved in 250 mL of THF. While a temp of 5°C was maintained, a mixture of 30 g (0.7 mol) of 95% formic acid and 30 mL of THF was added dropwise over 2 h. The resulting thick suspension was diluted with 520 mL of Et2O and stored at room temperature for 1 h. Filtration and washing with Et2O gave 122 g of (-)-norephedrine. Filtration and washing with Et2O gave 122 g of (-)-norephedrine formate, mp 144-145°C. The dried formate was refluxed for 2.5 days with 800 mL of toluene in a Dean-Stark trap until all the water was removed. The solvent was then evaporated, and vacuum drying gave 106 g (96%) of (-)-N-formylnorephedrine as a nd vacuum drying gave 106 g (96%) of (-)-N-formylnorephedrine as a slightly yellow syrup.
(-)-alpha-Amino-propiophenone Hydrochloride
The oxidation of (-)-N-Formylnorephedrine was carried out in four portions of 26.5 g with chromium trioxide-pyridine complex as described for the (+) antipode. The corresponding (-)-N-formyl-α-aminopropiophenone produced (91g, 83%) had the same NMR characteristics as (+)-N-formyl-α-aminopropiophenone. Hydrolysis of 90g (0.5 mol) of (-)-N-formyl-α-aminopropiophenone with 6 N hydrochloric acid (500 mL, 3 mol) at 40°C and evaporation of the excess acid provided (-)-α- Aminopropiophenone HCl as a tan solid, 65 g (69.5%). Two recrystallizations from i-PrOH-Et2O gave pure product (mp 175-176°C) which after vacuum drying over P2O5-KOH had a melting point of 188-190°C (dec). The salt weighed 56 g (60%). A mixture melting point with the other antipode or with racemic α-Aminopropiophenone gave a depression to 176-177°C dec.
Resolution of alpha-Aminopropiophenone With d-Tartaric Acid
A solution of 1.8 g (0.01 mol) of α-Aminopropiophenone HCl in 10 mL water was added all at once to an emulsion prepared by shaking 100 mL of CH2Cl2 and 10 mL of 1 M NaHCO3 solution. After the mixture was shaken for 20 s, the organic phase was separated as soon as possible and immediately added to a solution of 0.8 g (0.005 mol) of d- tartaric acid in 35 mL of EtOH. After 2 days at room temperature, 450 mg (17%) white crystals (mp 162-164°C (dec)), were separated. Treatment of the salt with ethanolic HCl gave (+)-α-Aminopropiophenone HCl: 280 mg (75%) from tartarate); mp 176°C.