Introduction
The Merck Index lists dimenoxadol as 'analgesic' with the remark 'abuse leads to habituation or addiction', which is an strong indication that it's a opiate type substance. Additionally it's a schedule I controlled substance.
Unlike in many other opiates, minor structural changes completely abolish the opiate activity. Substituents other than ethoxy or dimethylamine are not active, with one exception that will be mentioned latter.
Methods for the preparation can be found in Brit. Pat. 716,700, as well as brief information on the pharmacology. The preparation from benzilic acid is not as easy as one might think and involves rather unpleasant chemicals like sodium metal. It would most probably be better to acquire 2-dimethylaminoethyl benzilate, a spasmolytic, mydriatic, antirhinitic which is commercially available.
The two steps to Dimenoxadol are:
- Replacing the OH with a Cl, using thionyl chloride, and
- Forming ethyl ether by refluxing for 24h in absolute ethanol with CaCO3 added.
The LD50 in mice is 200 mg/kg sc and 650 mg/kg po, while the LD50 of morphine is 470-500 mg/kg sc and that of meperidine 195 mg/kg. Unlike the common opiates it supposedly does not suppress respiration, but has an atropine- and papaverinelike spasmolytic action. Even the hydrochloride is sparsely soluble in water (5-10%), making injections difficult. The potency is about 25% of morphine, i.e. the dosage is about 4 times higher. The recommended dosage is 1-3 mg/kg iv and 3-5 mg/kg im and sc. Alltogether, the pharmacological profile seems to resemble that of meperidine (pethidine).
An alternative compound is disclosed in Brit. Pat. 1,050,467. The preparation is similar to the one of dimenoxadol, but insted of ethanol there is 4-chlorobutanol (available commercially) used in the etherifying step. This compound has an activity slightly higher than morphine (+10%), with a LD50 of 220 mg/kg, therefore it's about 4 times safer and more potent than dimenoxadol. It also is a very powerful spasmolytic and anticonvulsant. Unlike morphine, it does not suppress, but rather increase respiration.
For the clandestine manufacturer (CM), this substance would have the advantage that it's not a controlled substance. One could also argue that it's not an analog of a controlled substance (dimenoxadol) but rather a derivative of a non-controlled substance (2-dimethylaminoethylbenzilate). This is an important point in countries with Controlled Substance Analogs laws. But better consult someone who knows more about CsA laws that me. One advantage for the CM is that none of the precursors are watched or suspicious chemicals.
However, the spasmolytic/anticholinergic activity could possibly spoil the fun with this substance, and it is not known if it produces a good high. There are other substances which can be made with similarely little effort and which are most likely much more rewarding. Other syntheses of Dimenoxadol have also been published5,6.
Procedure1
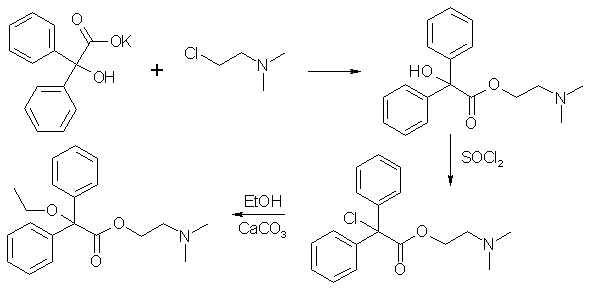
Method 1:
The sodium or potassium salt of benzilic acid was prepared, and reacted with 2-(dimethylamino)-ethyl chloride to form the benzilic acid ester, which was then reacted with thionyl chloride (SOCl2) to give diphenylchloroacetic acid 2-(dimethylamino)-ethyl ester hydrochloride.
35.5g (0.1 mol) of diphenylchloroacetic acid 2-(dimethylamino)-ethyl ester hydrochloride was dissolved in 400ml of absolute ethanol and stirred with 20g (0.2 mol) Calcium carbonate under reflux for 24h. The salts were filtered, the solvent evaporated in vacuum and the residue partitioned between 2N sodium carbonate and ether. The etheral solution was decolorized with activated carbon, filtered and extracted with 2N HCl until the extracts were clearly acid. The water was evaporated in vacuum, and the residue recrystallized from ethanol to give 70% of dimenoxadol hydrochloride, mp 166-167°C.
Method 2:
Diphenylchloroacetic acid was prepared by the action of thionyl chloride (SOCl2) on benzilic acid at room temperature.
23 grams of sodium metal were dissolved in 450 ml absolute ethanol and heated with 123g (0.5 mol) of diphenylchloroacetic acid on a water bath for 30 min, and the alcohol was distilled off in vacuo. The residue, dissolved in water, was shaken with ether, then acidified and again shaken with ether. The ether was evaporated and the residue, about 100g, was recrystallized from ethanol to give benzilic acid ethyl ether, mp 110-111°C (lit. 114°C)
25g of benzilic acid ethyl ether was boiled in 100ml acetone with 20g of potassium carbonate and 13g of 2-(dimethylamino)-ethyl chloride for 24h on a water bath. The acetone was distilled off and the residue partitioned between ether and water, the alkaline alkaline aqueous solution again shaken with ether, and the combined ether extracts extracted with 2N HCl until the extracts were clearly acid. The water was evaporated in vacuum, and the residue recrystallized from ethanol to give 65% of dimenoxadol hydrochloride, mp 165-6°C.
Synthesis of Benzilic Acid
Benzoin4
Dissolve 0.135 g of thiamine hydrochloride (Vitamin B1) in 0.27 mL deionized water in a 5-mL conical vial. Add 1.2 mL ethanol and cool the resulting solution in an ice bath. Slowly add 0.27 mL of cold 3 M sodium hydroxide dropwise over 2-3 min. Gently swirl the vial during the addition. Measure 0.75 mL of benzaldehyde and add it to the reaction mixture. Fit the vial with a condenser and reflux at ~60°C for 75-80 min or cap the reaction vial and allow it to stand for 48 h or more. Cool the reaction mixture to room temperature. Precipitate the benzoin by cooling the vial in an ice bath. Collect the solid by vacuum filtration and wash with cold water. Recrystallize the benzoin from ethanol.
Benzil3
Place 10g of powdered benzoin and 25ml of concentrated nitric acid in a 150ml flask fitted with a reflux water condensor, and heat the flask on a boiling water bath. A flask having a ground-glass neck fitting directly to the condensor is best for this purpose. If not available, fit the flask to the condensor by means of a cork (not a rubber stopper) and clamp both flask to the condensor securely in position during heating on the water bath: the nitrous fumes rot cork during the heating, and ifonly one clamp is used, the flask may possibly slip away from the condensor, or alternatively the latter may fall sideways under its own weight. Continue the heating for 1.5 hrs, when the crystalline benzoin will have been completely replaced by the oily benzil. Then pour the mixture into a beaker of cold water, when on vigorous stirring the oil will crystallise into a yellow solid. Filter off the later at the pump and wash thoroughly with water to ensure complete elimination of acid. Recrystallise from methylated or rectified spirit. Benzil separates as clear yellow crystals, mp 95°C yield 9g.
Benzilic Acid3
Dissolve 5g of benzil in 15ml of boiling ethanol in a conical flask fitted with a reflux water-condensor. Then add a solution of 5g of KOH in 10ml water, and heat the mixture (which rapidly develops a purple colour) on a boiling water-bath for about 15mins. Cool and stir the solution, from which the potassium benzilate separates in fine crystals. Filter the product at the pump, using an alkali-resisting filter-paper, or a sintered glass filter funnel. Wash the crystals on the filter with a small quantity of ethanol to remove the purple colour, and then drain thoroughly.
To obtain the free acid, dissolve the potassium salt in 50ml of cold water, filter the solution if a small undissolved residue remains; and boil the clear solution gently whilst dilute H2SO4 is added until the separation of the acid is complete. Cool the solution and filter off the pale orange-coloured crystals of the benzilic acid; wash the crystaks on the filter with some hot dH2O, drain well, and then dry in a dessicator. Yield of crude acid, 4g. Recrystallise from much hot water. The benzilic acid is obtained as colourless crystals, mp 150°C.