Abstract
Synthesis of the titled 2,5-dimethoxy-4-fluoroalkylamphetamine is reported. The highly functionalized aromatic nucleus of the key fluorination precursor was ultimately derived from a low temperature aromatic halogen-lithium exchange reaction followed by alkylation of the resultant anion with ethylene oxide.
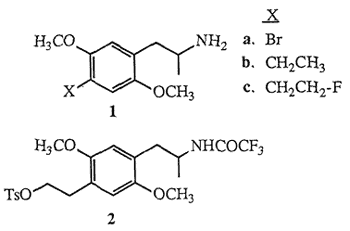
Molecules possessing the 2,5-dimethoxyphenyl-2- aminopropane (2,5-dimethoxyamphetamine) structural unit, which are appended with various substituents at the 4-position (para) of the aromatic nucleus are known to function as psychoactive agents in humans1. Included in the group of the most potent psychotomimetic amphetamines known are the para-substituted bromide 1a (DOB) and ethyl derivative 1b (DOET). The hypothesis that the mode-of-action of these and related para-substituted 2,5-dimethoxy- amphetamines involves the stimulation of the cerebral serotonin receptor system has been tested by evaluating their in vitro binding affinities toward the 5-HT2 and other serotonin receptor sub-types2. The potential in vivo assessment of serotonin receptor populations using the emerging technique of positron emission tomography3 (PET) provides impetus to further evaluate the structure-activity relationships of molecules related to DOET, in particular para-fluoroalkyl-2,5-dimethoxyamphetamines. Fluoroalkylamphetamines which exhibit favorable in vitro binding properties will permit evaluation of fluorine-18 (18F) radiolabeled ligands as potential PET imaging agents of the serotonin system.
The biological potency of DOET 1b coupled with the notion that fluorinated pharmaceuticals may elicit biological activities similar to the parent agents4 has prompted the synthesis of racemic 1-[2,5-dimethoxy- 4-(β-fluoroethyl)phenyl]-2-aminopropane (DOEF) 1c. In this communication we describe the synthesis of 1e employing a method which takes into account the constraints associated with the potential preparation of 18F-radiolabeled5 DOEF (18F-1c). The short half-life of fluorine-18 (110 min) would require a late synthetic introduction of the radiofluorine atom and also that subsequent chemical transformations be rapid, convenient and efficient. Based on these criteria tosyloxytrifluoroacetamide 2 was selected as the key intermediate for DOEF production. Initially, the preparation of the highly functionalized aromatic nucleus of 2 was envisaged to follow a synthetic route similar to the one used for the formation of DOET6, as outlined in Scheme I.
Scheme I
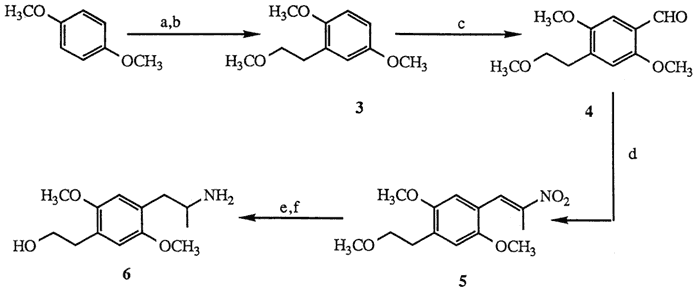
a. n-BuLi; ethylene oxide, 63%. b. NaH; CH3I, 88%. c. POCl3, HCON(CH3)C6H5, 24%. d. NH4OAc, CH3CH2NO2, 70%. e. AlH3, 93%. f. TMSCl, TMSI, 57%.
Treatment of 1,4-dimethoxybenzene with n-butyllithium7 (1.1 mol, THF, 0°C, then reflux 4.5 h) followed by ethylene oxide quench8 (1.1 mol, ether, 0°C; hexane, reflux 5 h, 63%) afforded the corresponding phenylethanol which was subsequently allowed to react with sodium hydride (1.0 mol, THF, reflux, 16 h) and then with iodomethane (1.1 mol, THF, 0°C,15 min; 20°C, 10 h, 88%) to provide methylether 39,10. Formylation of the para-position under Vilsmeier conditions6 (POCl3, HCON(CH3)C6H5, 10 min, 100°C, 24%) afforded para-benzaldehyde 4. Sidechain elaboration of 4 to nitropropene 511 (nitroethane 5 mol excess, cat. NH4OAc, 100°C, 2 h, 70 %) was followed by aluminum hydride reduction12 (5 mol LiAlH4, 100% H2SO4, THF, 0 °C; 20°C, 1 h; reflux 30 min, 95 %) to yield the corresponding aminoether. Deprotection of the methylethyl ether moiety by the method of Jung13 (TMSCl 2 mol, Et3N, CH2Cl2, 20°C, 30 min; then TMSI 1.3 mol, CHCl3, 20°C, 2 h, 57%) afforded aminoalcohol 6. The difficulty associated with the Vilsmeier transformation14 (3 to 4) precluded a useful large-scale production of 6, thus, an alternate synthesis was devised according to the route shown in Scheme II.
Scheme II
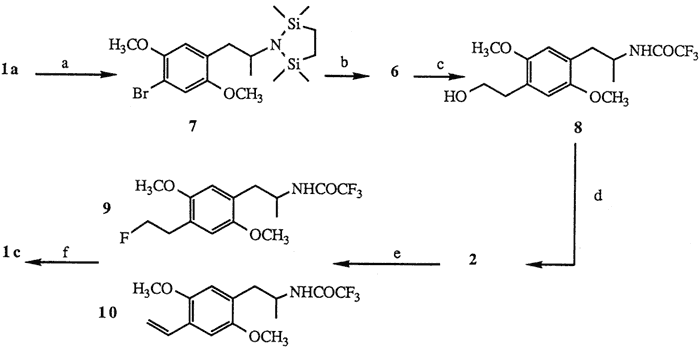
a. [ClSi(CH3)2CH2]2, 85%. b. tert-BuLi; ethylene oxide; KOH, 75%. c. (CF3CO)2O; NaHCO3, 89%. d. TsCl, 96%. e. TBAF, 55%. f. KOH, 86%.
The facile synthesis of 6 was effected by employing the readily available starting material DOB15, Protection of 1a using Magnus' reagent16 ([ClSi(CH3)2CH2]2 1 mol, Et3N, CH2Cl2, 20°C, 2 h, 85%) yielded bromodisilazane 7. Low temperature treatment of 7 with tert-butyllithium7,17 (2.1 mol, ether, -78°C, 15 min) followed by addition of ethylene oxide (1.5 mol, Et2O, -78°C, 1 h; then hexane, reflux, 3 h) and finally liberation of the amine functionality under basic conditions16 (10% KOH, MeOH, reflux, 4 h) provided aminoalcohol 6 (75% based on 7). Subsequent formation of hydroxyacetamide 8 was carried out with a two step procedure18 ((CF3CO)2O 2.5 mol, Et3N, Et2O, 0°C; 35°C, 1 h; then NaHCO3 1 mol, MeOH, H2O, 20°C, 3.5 h, 89%).
Subjection of 8 to standard tosylation conditions (p-TsCl, py, 0°C, 12 h, 96%) provided the desired tosylate 2. Of the many sources of fluoride ion reported to displace sulfonate esters19 treatment of 2 with a solution of commercially available tetrabutylammonium fluoride (TBAF)20 (1.5 mol, THF, reflux, 30 min) afforded after separation (HPLC) fluorinated amide 9 (55%) and styrene 1021 (18%). Subsequent hydrolysis of the trifluoroacetamide protecting group of 9 (KOH 1 mol, H2O, i-PrOH, 45°C, 30 min, 86%) yielded fluoroamphetamine 1c.
In summary, the synthesis of 1e was achieved in 8 steps starting from DOB in an overall yield of 26%. The critical transformation for the synthesis of key intermediate 2 involved a low temperature halogen-lithium exchange reaction of 7 followed by alkylation of the resultant anion with ethylene oxide. The method undoubtedly will prove valuable for the construction of other molecules containing the 1,4-dialkyl-2,5-dimethoxybenzene nucleus. Furthermore, the expedient (<75 min) and facile (47%) two-step transformation of tosylate 2 to DOEF is noteworthy, well-suited for the production of radiolabeled 18F-1c and provides a general route to other fluoroalkylaromatic pharmaceuticals. The results of the in vitro receptor binding affinity of 1c toward the 5-HT2 serotonin receptor and also the radiosynthesis of 18F-1c will be reported elsewhere.