We have been studying synthetic pathways to 5-substituted analogs of Melatonin a pineal gland hormone which has been implicated1,2 in many physiological and Psychological problems. The facile preparation of the precursor 5-substituted indoles is of importance in its own right as many of these compounds are in clinical use3. The reaction pathway discussed is a major variation of the "Indole-Indoline-Indole" synthetic sequence. In 1962, Thesing, Semler and Mohr4 described the reaction of indole (I) with aqueous ethanolic sodium bisulfite to produce sodium indoline-2-sulfonate This salt, being a substituted aniline, undergoes electrophilic substitution para (5-position of the indole ring) to the ring nitrogen whereas indole undergoes substitution primarily at the 3-position5: treatment with base regenerates the substituted indole. Although this reaction should be the beginning of a powerful approach to the 5-substituted indoles, the low reported yields, lack of definitive characterization of the intermediates and the paucity of reported examples (the only subsequent reported use was a failure6), led us to a detailed examination of the sequence shown in Scheme 1.
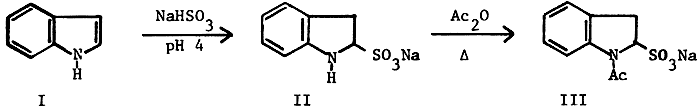
Thesing reported obtaining II in 68% yield but presented no spectral data or analytical evidence for the nature of the salt other than limited my curves. The compound was unstable toward heat or water and reverted to indole. We have succeeded in showing that II is isolated as a monohydrate. Instead of a single infrared band in the amine region, two relatively sharp bands were observed, one at 3290 cm-1 for the N-H stretch and another at 3490 cm-1 which was assigned to the water of hydration. The elemental analysis of II confirms the hydrate formulation. The presence of water in the crystal lattice and the ready hydrolysis of II back to indole with no other source of water present was confirmed by an NMR study. The spectrum of II in DMSO-d6 shows a signal at 3.30 due to the water of hydration. A series of spectra performed on a sample which was allowed to remain in the probe (about 35°C) over a 40 hr period showed radical changes after 16 hrs. Spectra run at 24, 32 and 40 hrs demonstrated the gradual reversion to indole with the 40 hr spectrum being virtually identical to that of indole in DMSO-d6. Based on a detailed comparison plot of over 22 experimental runs, a combination of longer stir time, less ethanol, more water and slightly increased bisulfite usage resulted in yields consistently greater than 97%. Five larger scale (1-3 moles) preparations have all given yields of II in excess of 98%.
The subsequent acetylation also proved to be an unexpected problem. The literature procedure4 used the crude, wet product without purification and there was no spectral or analytical data about the exact nature of the compound. We have isolated the dry sodium 1-acetylindoline-2-sulfonate (III), as a white solid which has displayed an infrared band showing the presence of water even after drying in vacuo at 110°C; the elemental analysis confirmed it as a hemihydrate. Here again, a brief study of the reaction parameters and changes made based on the information obtained resulted in an 85% yield of III; larger scale runs averaged between 82-85%. These results have meant correspondingly higher yields of the 5-substituted indoles shown in Scheme 2.
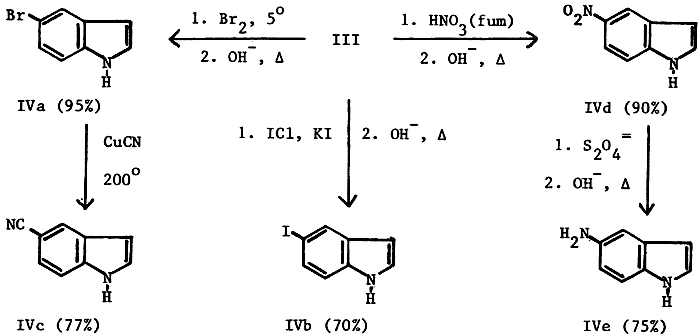
While 5-bromoindole (IVa), 5-iodoindole (IVb) and 5-cyanoindole (IVc) were obtained by relatively simple procedures, the preparation of 5-nitroindole (IVd) and of 5- aminoindole (IVe) was somewhat more complex. Four different, relatively standard nitration procedures gave yields of only 40%. A new procedure by Crivello7 failed probably because of the insolubility of III in the required solvent. Fuming nitric acid in glacial acetic acid, at the freezing point of acetic acid, proved to be superior for the nitration of III, affording consistent yields of 90%.
The catalytic reduction of IVd8,9 seems useful for only relatively small (1-20 g) quantities of nitro compound. Since approximately 500g was required for future work, a number of standard, acidic reduction procedures applicable to larger scale reactions were investigated; however, these gave only poor to moderate yields. A possible reason for these reduced yields may be dimerization and trimerization of the relatively electron-rich aminoindole product at the 2- and 3-positions. In order to circumvent this possibility we investigated reduction under basic conditions and found that the use of sodium hydrosulfite in 1 N sodium hydroxide was the most successful10 and resulted in reproducible yields of over 75% on a 0.4 mole scale.
Table 1.
| Method | 5-Bromo |
5-Iodo |
5-Cyano |
5-Nitro |
5-Amino |
| From Indoline | 34% |
30% |
28% |
50% |
41% |
| Thesing4 | 48% |
40% |
28% |
26% |
21% |
| Present work | 80% |
57% |
62% |
74% |
57% |
The overall results of our work are compared in Table 1 to those of Thesing et al.4 and to those of the best composite literature results starting from indoline.
Our results suggest that this sequence should be of major consideration for the preparation of 5-substituted indoles. To our knowledge there have been no reports of the bisulfite reaction with indoles substituted at the 1, 2 and/or 3 positions and these potentially significant reactions are currently under investigation in our laboratory.
Experimental
Sodium Indoline-2-sulfonate Monohydrate (II)
To a solution of 23.4 g of sodium bisulfite (59% as SO2) in 80 ml of water was poured slowly, with stirring, a solution of 11.7 g (0.1 mole) of indole in 25 ml of ethanol. The resulting Yellowish mixture was stirred for 20 hrs at room temperature and passed through various color changes. The final thick, slightly green slurry was filtered by suction; the solid was then washed with 25 ml of methanol two 50 ml portions of ether and air dried to yield 23.7 g (98%) of white solid which decomposed between 340 and 360°C.
Sodium 1-Acetylindoline-2-sulfonate Hemihydrate (III)
A slurry of 23.9g (0.1 mole) of the salt II in 150 ml of acetic anhydride was heated to 70°C and was maintained at this temperature for 2 hrs with stirring. The temperature was then raised to 90°C for 0.5 hr during which time the very thick slurry slowly thinned and turned a pale tan color. The reaction mixture was cooled to 200 and the solid was collected. The filter cake was washed with three 40 ml portions of ether and air-dried overnight. Further drying at 110°C (oven) gave 22.8 g (83%) of white powder which decomposed above 300°C.
5-Bromoindole (IVa)
A solution of 240 g (0.88 mole) of III in 1100 ml of water was cooled to 0-5°C. To this was added, dropwise with stirring, 155g (0.97 mole) of bromine at such a rate as to keep the temperature below 5°C. After the addition was complete, the deep orange slurry was stirred at 0°C for 1 hr and allowed to warm to room temperature over a 1 hr period. The solution was diluted with 1.5 L of water and sodium bisulfite was added to eliminate any excess bromine. To the now faint yellow reaction was added 150 g of solid sodium hydroxide and reflux was continued overnight (20 hrs). The mixture was cooled to 10, the solid was collected and washed with three 500 ml portions of cold water and dried overnight at 700 to give 159.5g (92%) of tan crystals, mp 88-90°C, lit4,9 90-91°C; its infrared spectrum was identical to that of an authentic sample (Aldrich, mp 90-92°C). An additional 10.5 g (0.054 mole) of tan product, mp 87-89°C, was obtained by ether extraction of the filtrate, thus making the total yield 98%. 5-Iodoindole (IVb) To a solution of 14.1 g (0.052 mole) of III and 10 g (0.06 mole) of potassium iodide in 60 ml of water, cooled to 0-5°C, was added dropwise over a 1 hr period iodine monochloride (24.4 g, 7.6 ml, 0.15 mole); the temperature was maintained below 5°C. The deep maroon slurry was allowed to warm to room temperature, poured into 200 ml of water with stirring, the flask rinsed with 50 ml of water and a small amount of sodium bisulfite added to destroy any excess iodine. The faint yellow solution was neutralized with 20% sodium hydroxide, 6 g of solid sodium hydroxide was added and the mixture refluxed overnight (20 hrs). The reaction mixture was cooled to 10°C, the solid collected and washed with three 25 ml portions of ice water and air dried overnight to give 8.1 g (65%) of tan solid, mp 97-100°C, lit4,6 99-102°C.
5-Cyanoindole (IVc)
A mixture of 168.0 g (0.86 mole) of IVa, 650 ml of N-methyl-Pyrrolidinone and 120 g (1.35 moles) of cuprous cyanide were boiled under reflux (200°C) for 18 hrs. After cooling to room temperature the reaction mixture was poured into 2 L of ice water, stirred for 0.5 hr and collected. The filter cake was slurried in 3 L of 20% aqueous ammonia, filtered through sintered glass, washed with five 100 ml portions of 20% aqueous ammonia until the washings were a faint blue and then washed with 500 ml of ice water. The cake was dried and the crude product was extracted continuously with hot chloroform (Soxhlet) for 20 hrs. The solvent was evaporated to give 95.3 g (18%) of tan solid, mp 104-106°C, lit4,9 107°C. The infrared spectrum was identical to that of an authentic sample (Aldrich, mp 106-108°C).
5-Nitroindole (IVd)
Compound III (272.0g, 1.0 mole) was dissolved in 2 L of glacial acetic acid with stirring and cooled to 12-15°C. Fuming nitric acid (190 ml, 90%, 2.4 mole was added dropwise over about 1 hr with the temperature riding between 12-15°C. Water (2500 ml), crushed ice (1500 g) and a total of 1600 g of solid sodium hydroxide were added in portions with the temperature slowly rising to 65-70°C The resulting thin yellow slurry thickened considerably after stirring overnight (20 hrs). Collection, washing with two 200 ml portions of ice water and drying gave 145.5g (90%) of a yellow solid, mp 134-137°C, lit4 140-141°C. The infrared spectrum was identical to that of an authentic sample (Aldrich mp 134-137°C)
5-Aminoindole (IVe)
The identification and characterization of this compound is complicated by relatively facile air oxidation when in solution. Numerous literature mp's range from 126°C to 135°C8,9,11 The melting point of the Aldrich material is 131-135°C.
- A. With Iron/HCl in Ethanol
- A solution of 8.1 g (0.05 mole) of IVd was dissolved in 500 ml of ethanol (minimum required) at 55-60°C. Iron powder (6.2 g, 0.11 mole) was added with vigorous stirring and 3 ml of conc. hydrochloric acid was added cautiously over a 15 min. period. This mixture was allowed to stir for 4 hrs at 78-80°C. It was filtered hot and the black cake washed with 50 ml of hot ethanol. The solvent was evaporated on a rotary evaporator to give 3.2 g (49%) of brown solid mp 128-132°C. The IR (KBr) spectrum showed N-H bands at 3400, 3340 and 3200 cm-1 but the cornpound was obviously impure.
- B. With Iron/Acetic Acid
- Glacial acetic acid (15 ml) was added to 8.1 g (0.05 mole) of IVd and the mixture heated to 60°C. Iron powder 6.9 g, 0.12 mole) was added in small increments over a period of 4-5 min and the mixture vigorously stirred at 60° for 1 hr. The reaction mixture was cooled to room temperature and made alkaline with 20% sodium hydroxide. The resulting greenish slurry was filtered (very slow) and the filtrate extracted with three 100 ml portions of ether. The extracts were dried (sodium sulfate) and evaporatedd to yield 3.5 g (40%) of light purple solid, mp 128-132°C. The infrared spectrum matched that of an authentic sample. With Sodium Hydrosulfite a solution of 8.1 g (0.05 mole) of IVd in 200 ml of ethanol was heated to 55°C with stirring after 1 N sodium hydroxide (200 ml) had been added. A solution of 43.5g (0.25 mole) of sodium hydrosulfite in 200 ml of 1 N sodium hydroxide was added dropwise over a period of 15 min and the resulting mixture allowed to stir for 24 hr at 80°C. The reaction was filtered hot, concentrated and the precipitate in the cooled, aqueous portion filtered and dried to give 3.3 g (0.025 mole) of pale violet solid, mp 128-130°C. Extraction of the filtrate with three 100 ml portions of ether, drying and evaporation of the combined extracts gave an additional 1.7 g (0.013 mole) of slightly darker purple crystals, mp. 129-132°C. The infrared spectra of the above fractions were identical to that of an authentic sample (Aldrich). This represents a total yield of 76%. A repeat of this procedure on a 0.4 mole scale gave a 78% yield.