Abstract
The polymethylhydrosiloxane-B(C6F5)3 combination is found to be a versatile carbonyl defunctionalization system under mild and rapid conditions. For the first time, B(C6F5)3 has been used as a nonconventional Lewis acid catalyst to activate PMHS. Aromatic and aliphatic carbonyl compounds were effectively reduced to give the corresponding alkanes in high yields.
Defunctionalization of organic functional groups is an equally desirable achievement as compared to functionalization. There is a great need to discover new methodologies for defunctionalization especially for conversion of polyfunctional natural products to useful building blocks and bioactive molecules. Available literature speaks of only a few protocols for removal of a certain functional group, viz., the carbonyl group can be defunctionalized to a methylene group by Clemmensen1 or Wolff-Kishner reduction,2 both of which require very drastic reaction conditions. The hydroxyl group can be removed by a Barton-McCombie procedure,3 wherein highly malodorous xanthate and Bu3SnH are required. Some other methods known in the literature include catalytic hydrogenation4 and reaction involving use of PtO2,5 HI-phosphorus,6 BH3,7 Zn/HCl/HgCl2/H2O,8 NaBH4-CF3CO2H,9 NaCNBH3-BF3 Et2O,10 LAH-AlCl3,11 Et3SiH-BF3·Et2O or CF3CO2H12 besides a few others.13 The majority of the known procedures used for defunctionalization are nonchemoselective and require harsh reaction conditions.
All of these methods, while offering some advantages, also suffer from disadvantages. Most of these methods are generally restricted to aromatic systems, are some times harsh and need a pyrophoric hydride source for reduction, require longer reaction hours with careful workup procedures for quenching the excess reagent, and are often associated with low yields. The usefulness of polymeric hydride source polymethylhydrosiloxane (PMHS), a coproduct of the silicone industry, as an excellent reduction reagent is well demonstrated in several recent publications.14,15 The quest to find newer activators for this rather inert polymer resulted in identification of tris(pentafluorophenyl)borane as an excellent catalyst for activation of PMHS. B(C6F5)316 is a relatively unexplored Lewis acid. This combination of PMHS-B(C6F5)3 is found to be a versatile carbonyl defunctionalization system with very short reaction times (Scheme 1). Interestingly, this combination establishes a powerful "catalytic switch", viz., our initial studies using ZnCl2 as an activator resulted in reduction of ketone to alcohol,17 whereas this new catalyst promoted the reduction of the same substrate to methylene group.18 The procedure is very simple, and the reaction completion is indicated by the termination of effervescence (reaction times ranging from 5 to 20 min).
Scheme 1
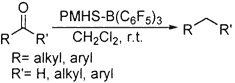
The earlier procedures involving PMHS as a hydride source for similar transformations required stoichiometric amounts of AlCl3 as an activator13d and were limited to aryl carbonyl compounds, whereas in the case of Pd/C as an activator,13f double bonds are reduced to saturation instead of carbonyl reduction to a methylene group.
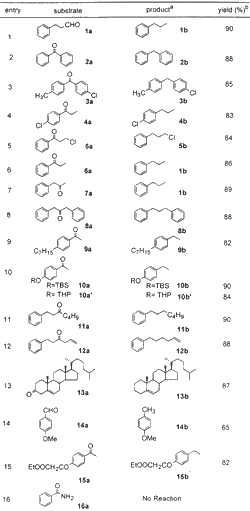
Table 1
Reduction of Carbonyl Compounds
Notes
- All products characterized
by 1H NMR and MS - Isolated yields.
To establish the optimum reaction conditions, the reaction was first studied on readily available benzophenone 2a, which was reduced to diphenylmethane 2b in 88% isolated yield (entry 2) in 10 min. Another substrate, phenyl propanaldehyde 1a, was reduced to n-propyl benzene 1b in 90% yield (entry 1) in 8 min. These two examples demonstrate that not only benzylic ketone (a very easily reducible carbonyl) but also aliphatic aldehyde is suitable for the present protocol (Table 1). The halo-substituted aryl ketones 3a, 4a, and 5a also were reduced to the corresponding methylene compounds 3b, 4b, and 5b without affecting the aryl halide group (entries 3 and 4). The alkyl halide group (entry 5) is also unaffected under the present protocol. Aliphatic keto substrates 7a, 8a, 11a, 12a, and 13a were also well suited to the present reaction conditions (entries 7, 8, and 11-13). Substrates 10a and 10a' demonstrate the selective reduction of ketone in the presence of TBS and THP ethers (entry 10), and substrate 12a demonstrates inertness to olefin functionality (entry 12). The steroid substrate 13a was reduced without isomerization of double bond in 87% yield requiring only 10 min (entry 13). The reduction of anisaldehyde 14a to 4-methyl anisole 14b was also achieved, albeit in low yield (65%). In the case of compound 15a (entry 15), where both carbonyl and ester groups are present in the same substrate, selective reduction of carbonyl group to methylene is observed in 82% isolated yield. Attempts, however, to reduce benzamide 16a to benzylamine (entry 16) were futile.
Scheme 2
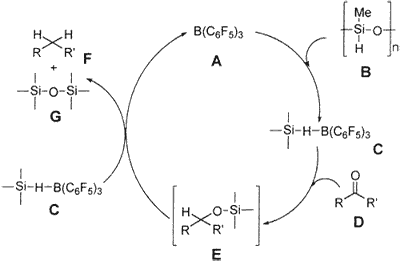
Hypothetically, we propose that complex C, which is formed from B(C6F5)3 A and PMHS B, is responsible for the reduction of the carbonyl functionality.16a Complex C would react with carbonyl group D to form E (not isolated), which would produce the reaction product, hydrocarbon F, and silyl ether G and would regenerate A (Scheme 2). Coordination of carbonyl oxygen to boron for facile hydride transfer is also expected. There is literature precedence that Ph3SiH and Et3SiH also operate in a more or less similar pathway; the intermediate E is isolable when 1 equiv of Ph3SiH is used,16b and longer reaction times (20 h) are required when Et3SiH is used.16c However, in the case of PMHS, even though 1 equiv of reagent is used, the intermediate E could not be isolated, and instead 45-50% hydrocarbon conversion was observed within 5-20 min and the remaining starting carbonyl was isolated. This clearly indicates that PMHS is a more powerful reducing agent than Ph3SiH and Et3SiH in the presence of B(C6F5)3.
In Summary, we have demonstrated for the first time, a direct and rapid conversion of carbonyl functionality to methylene group under very mild conditions with high yields. The procedure is very simple, and progress of the reaction can be monitored by visualization without any analytical support. The shorter reaction time in all the cases studied is an added advantage.
Experimental
General Methods
Silica gel used was 60-120 mesh. 1H NMR spectra were obtained in CDCl3 at 200 MHz. Chemical shifts are given in parts per million with respect to internal TMS, and J values are given in hertz. Methylene chloride was distilled over CaH2 prior to use.
General Procedure for Defunctionalization of Carbonyl Group with PMHS-B(C6F5)3
To a solution of carbonyl compound (1 mmol) in dry CH2Cl2 (5 mL) and tris(pentafluorophenyl)borane (5 mol%) was slowly added polymethylhydrosiloxane (3 mmol) at room temperature. After 5-20 min, a vigorous effervescence (like foam) was observed. At this point, the solvent was evaporated and reaction mixture was dissolved in hexane and filtered through a silica gel pad using hexane. Evaporation of the volatiles afforded the reduction product in pure form. Spectral data of all products other than 9b, 10b, and 10b' were identical with those of authentic samples.19