Abstract
The nitro group in aliphatic and aromatic nitro compounds, which also contain reducible substituents such as alkenes, nitriles, carboxylic acids, phenols, halogens, esters, etc., is selectively and rapidly reduced at room temperature to the corresponding amine in good yield by employing hydrazinium monoformate in the presence of magnesium powder. It was observed that, hydrazinium monoformate is more effective than hydrazine or formic acid or ammonium formate and reduction of the nitro group occurs without hydrogenolysis in the presence of low-cost magnesium compared to expensive metals like palladium, platinum, ruthenium, etc.
Introduction
Rapid and selective reduction of nitro compounds is of importance for the preparation of amines, particularly when a molecule contains other reducible moieties1-5. The synthesis and biological evaluation of aromatic amines are active and important areas of research and their chemistry by derivative formation has been widely studied6-7. Numerous new reagents have been developed for the reduction of aromatic nitro compounds8-9. Though some of these are widely used, they still have limitations based on safety and handling considerations10-12. To overcome these difficulties, several new methodologies have been developed13-14. However, little attention has been paid to the reduction of aliphatic nitro compounds15-17, which traditionally are reduced by high-pressure catalytic hydrogenation18-20. Many of these reduction methods also have substantial limitations with regard to chemoselectivity. For example, poor selectivity was reported in the reduction of aromatic nitro compounds, which have halogens, nitrile, carboxyl, hydroxyl, etc., as substituents. Reduction at reflux temperature11,21 for several hours can cause rearrangements and cyclization in polyfunctional nitro compounds. Therefore, we examined several methods to improve the reduction process, and, especially, to obtain selectivity over reducible or other labile substituents. In this context, the use of 5% platinum on carbon was found to be efficient but not cost effective22.
Results And Discussion
In this communication, we wish to report, a rapid and simple reduction of aliphatic and aromatic nitro compounds to the corresponding amino derivatives by using magnesium powder and hydrazinium monoformate, at room temperature [Eq. 1].
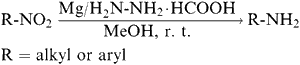
The reduction of nitro compounds in the presence of magnesium powder and hydrazinium monoformate is completed within one to ten minutes with the course of the reaction monitored by thin layer chromatography and IR spectra. No bands are observed for -N=N-, -NH-OH, -N=O moieties. The work-up and isolation of the products were easy and all the compounds reduced (Table 1) by this system were obtained in good yields (90ñ95%). The products were characterized by comparison of their TLC, IR spectra and melting points with authentic samples. Control experiments were carried out using a few of the nitro compounds with hydrazinium monoformate, but without magnesium powder, did not yield the desired products. In order to test the selectivity, reductions were attempted with p-dichlorobenzene, p-chloro-m-cresol, β-naphthol, cinnamic acid, acetanilide, benzoic acid, anisole, benzonitrile and phenyl acetate at laboratory temperature. However, these reactions failed to give any reduced product.
Table I
Magnesium Catalyzed Reduction of Nitro Compounds Using Hydrazinium Monoformate
| Nitro Compound | Reaction Time |
Product | Yielda |
Melting Point (∞C) |
|
Found |
Lit. |
||||
| o-Nitrophenol | 2.5 min | o-Aminophenol | 94% |
173ñ175 | 174 |
| m-Nitrophenol | 2 min | m-Aminophenol | 95% |
121ñ123 | 123 |
| o-Nitrotoluene | 3 min | o-Toluidineb | 92% |
142ñ144 | 144 |
| m-Nitrotoluene | 2 min | m-Toluidineb | 93% |
124ñ126 | 125 |
| p-Nitrotoluene | 5 min | p-Toluidine | 92% |
44ñ45 | 45 |
| α-Nitronaphthalene | 2 min | α-Naphthylamine | 92% |
50ñ51 | 50 |
| β-Nitronaphthalene | 2 min | β-Naphthylamine | 94% |
111ñ113 | 113 |
| o-Nitroanisole | 3 min | o-Anisidineb | 94% |
58ñ60 | 60 |
| m-Nitroanisole | 2 min | m-Anisidinec | 95% |
81ñ82 | 80 |
| p-Nitroanisole | 3 min | p-Anisidine | 95% |
56ñ57 | 57 |
| o-Nitroaniline | 2.5 min | o-Phenylenediamine | 93% |
100ñ103 | 102 |
| m-Nitroaniline | 2 min | m-Phenylenediamine | 94% |
64ñ65 | 64 |
| p-Nitroaniline | 3 min | p-Phenylenediamine | 94% |
140ñ143 | 141 |
| m-Nitrobenzyl alcohol | 3 min | m-Aminobenzyl alcohol | 91% |
96ñ98 | 97 |
| p-Nitrobenzamide | 2.5 min | p-Aminobenzamide | 92% |
115ñ116 | 114 |
| p-Nitrophenylacetate | 2.5 min | p-Aminophenylacetatec | 93% |
148ñ151 | 150 |
| o-Nitrobenzoic acid | 5 min | o-Aminobenzoic acid | 85% |
144ñ147 | 145 |
| o-Nitrochlorobenzene | 0.5 min | o-Chloroanilineb | 93% |
99ñ100 | 99 |
| m-Nitrochlorobenzene | 0.5 min | m-Chloroanilineb | 91% |
120ñ123 | 122 |
| p- Nitrochlorobenzene | 0.5 min | p-Chloroaniline | 93% |
70ñ71 | 71 |
| o-Nitrobromobenzene | 1 min | o-Bromoanilineb | 94% |
115ñ117 | 116 |
| m-Nitrobromobenzene | 1 min | m-Bromoanilineb | 94% |
118ñ121 | 120 |
| p-Nitrobromobenzene | 1 min | p-Bromoaniline | 93% |
65ñ66 | 66 |
| m-Nitroiodobenzene | 2 min | m-Iodoaniline | 92% |
119ñ121 | 119 |
| p-Nitrocinnamic acid | 3 min | p-Aminocinnamic acidd | 90% |
265ñ268 | 265ñ270 |
| p-Nitrobenzonitrile | 2.5 min | p-Aminobenzonitrile | 92% |
84ñ85 | 83ñ85 |
| p-Nitrophenylacetonitrile | 2 min | p-Aminophenylacetonitrile | 91% |
45ñ48 | 45ñ48 |
| p-Nitrophenethyl alcohol | 2 min | p-Aminophenethyl alcohol | 90% |
108ñ111 | 108ñ110 |
| p-Nitroacetanilide | 2 min | p-Aminoacetanilide | 94% |
163ñ165 | 163 |
| Methyl p-nitrocinnamate | 3 min | Methyl p-aminocinnamate | 90% |
128ñ130 | 129 |
| Nitromethane | 1 min | Methylamined | 75% |
230ñ233 | 232ñ234 |
| Nitroethane | 1 min |
Ethylamined | 77% |
106ñ108 | 107ñ108 |
| 1-Nitropropane | 1.5 min |
1-Aminopropaned | 80% |
158ñ160 | 160ñ162 |
| 1-Nitrobutane | 2 min |
1-Aminobutanee | 74% |
78ñ80 | 78 |
- Isolated yields are based on a single experiment and the yields were not optimized.
- Isolated as benzoyl derivative.
- Isolated as acetyl derivative.
- Isolated as hydrochloride salt.
- Boiling point.
Several interesting features of the reduction reaction are apparent from Table I. First, electron-withdrawing substituents enhance the rate of reduction. The reduction of nitro compounds having electron-withdrawing substituents was completed within 1 min. On the other hand, electron-releasing groups slow the reaction rate. The degree of variation of reaction time depends on the position of the electron releasing substituents. Thus m-nitroaniline undergoes faster reduction than o-nitroaniline. The latter is reduced faster than p-nitroaniline.
Summary and Outlook
In summary, magnesium/hydrazinium monoformate provides a convenient system for the selective reduction of aliphatic and aromatic nitro compounds. The source of hydrogen for this catalytic transfer hydrogenation method is hydrazinium monoformate, a new hydrogen donor, which is inexpensive, more effective than either hydrazine or formic acid. Most of the reactions were completed within 2 min as monitored by the disappearance of the starting materials and concomitant formation of the product (TLC). This system is not helpful in obtaining directly an amino carbonyl compound, due to the formation of hydrazone derivatives with the donor.
Thus, the reduction of nitro compounds can be accomplished with magnesium powder instead of expensive platinum or palladium, etc., without affecting the other reducible or hydrogenolysable substituents. The yields are virtually quantitative and analytically pure products are obtained. The obvious advantages of the proposed method over previous methods are: (i) selective reduction of nitro compounds in the presence of other reducible or hydrogenolysable groups, (ii) easy procedure, (iii) rapid reduction, (iv) high yields of substituted amines, (v) avoidance of strong acid media, (vi) no requirement for pressure apparatus and (vii) inexpensive. This procedure, therefore, will be of general use, especially in cases where rapid, mild and selective reduction is required. Further investigations of other useful applications related to deblocking of protecting groups in peptide synthesis are in progress.
Experimental
Materials
All of the nitro compounds, hydrazine hydrate and formic acid were purchased from Aldrich Chemical Company (USA). The nitro compounds were recrystallized or distilled prior to use. Magnesium powder and 60-120-mesh silica gel (for column chromatography) were purchased from SISCO Research Laboratories Pvt. Ltd., Bombay (India). The magnesium was treated with 0.01N hydrochloric acid for about 2 min. It was then filtered through a sintered glass funnel and washed with water, dry methanol and dry ether. The thus obtained magnesium was vacuum dried and stored. All of the solvents used were analytical grade or were purified according to standard procedures. Thin layer chromatography was carried out on silica gel plates obtained from Whatman, Inc. The melting points were determined by a ThomasñHoover melting point apparatus and are uncorrected. IR spectra were recorded on a Shimadzu FTIR-8300 spectrometer.
Typical Procedure
Hydrazinium monoformate was prepared by neutralizing slowly, equal moles of hydrazine hydrate and 85% formic acid (no solvent) in an ice water bath, with constant stirring. The obtained hydrazinium monoformate solution was used as such for the reductions. A suspension of an appropriate nitro compound (5 mmol) and magnesium powder (10 mmol) in methanol or in any other suitable solvent (5 mL) was stirred under nitrogen atmosphere with hydrazinium monoformate (2 mL) at room temperature. After the completion of the reaction (monitored by TLC), the catalyst was filtered off. The residue was extracted with 15 mL of chloroform or dichloromethane or diethyl ether. The extract was washed twice with 15 mL saturated sodium chloride solution and then with 10 mL water. The organic layer was dried (Na2SO4) and then evaporated to obtain the desired amino derivative.
In order to obtain good yield of volatile aliphatic amine, the reaction was carried out by the controlled addition of hydrazinium monoformate through a condenser cooled with ice water and by immersing the reaction flask in a cold-water bath. After filtration, the reaction mixture was neutralized with HCl. The solvent was evaporated under reduced pressure. The residue was lyophilized or subjected to column chromatography by using 60-120 mesh silica gel and a suitable eluting system (50:50 chloroform:benzene, 60:40 chloroform:benzene, 80:20 chloroform:benzene, 90:10 chloroform:benzene, 80:20 chloroform:methanol, 85:15 chloroform:methanol, 90:10 chloroform:methanol, 95:5 chloroform:methanol). Aliphatic amines were obtained as their hydrochloride salts in up to 80% yields.