Abstract
Various oximes, both aldoximes and ketoximes, are selectively reduced to corresponding amines employing low cost zinc dust and ammonium formate despite presence of other functional groups such as halogens, -OH, -OCH3, -COOH, -CN, >C=C< and -CH3.
The conversion of carbonyl derivatives to amines via oximes is a useful transformation in the synthesis of numerous organic compounds and also during the synthesis of compounds which are key intermediates in biosynthesis of many pharmacological important substances. Numerous new reagents have been developed for the reduction of oximes to amines.1-8 Though some of these are widely used, still they have limitations based on chemoselectivity and economic considerations.
The heterogeneous catalytic transfer hydrogenation method has proved more effective for reduction of organic compounds than traditional hydrogenation or other methods of reduction, as it involves mild reaction conditions, easy work-up and a high degree of selectivity9-12. Earlier reports reveal that catalytic transfer hydrogenation of oximes to amines had been achieved with systems like ammonium formate/10% Pd/C13, cyclohexene/10% Pd/C14, but these systems require reaction times as long as 5-10 hours at reflux, expensive catalysts and also offer very low yields. Moreover, stringent precautions must be taken while employing palladium on carbon because of its flammable nature in presence of air.
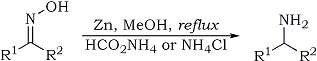
Scheme 1
R1, R2 = H, alkyl, phenyl or substituted phenyl group
In this communication, we report a rapid, selective and simple reduction of oximes to corresponding primary amines using low cost zinc dust and ammonium formate or ammonium chloride at reflux temperature as depicted in Scheme 1. Various other functionalities like halogens, -OH, -OCH3, -COOH, -CN, >C=C< and -CH3 are tolerated. Further, we observed that in the case of nitro oximes, the nitro group on the aryl residue underwent reduction to the amine function at room temperature with this system, whereas the oxime function undergoes reduction only at reflux temperature to yield the corresponding diamine.
The reduction of oximes in the presence of zinc dust and ammonium formate was completed within two to five minutes. The course of reaction was monitored by TLC and IR spectra. The work-up and isolation of the products were easy. Thus, the oximes reduced (a few examples are listed in Table 1) by this system were obtained in good yields (90-95%). The products were characterised by comparison of their boiling points or melting points, TLC and IR spectra with authentic samples. Further characterisation of the products was done by converting them to known derivatives and also by elementary analysis. The disappearance of strong absorption bands between 1690 and 1640 cm-1 due to C=N stretching and between 3650 and 3500cm-1 due to O-H stretching and appearance of two strong absorption bands between 3500 and 3300 cm-1 of -NH2 group clearly show that the oximes were reduced to the corresponding primary amines.
The use of ammonium chloride for the reduction of nitro compounds to amines15 provoked us to investigate the reduction reaction by replacing ammonium formate by ammonium chloride, which performs the conversion of oximes to amines at a slow rate. The complete conversion requires at least 2-3 hours at reflux temperature. This may be due to the fact that the solubility of ammonium chloride is very poor compared to ammonium formate and also in the case of ammonium formate, formate ion also provides hydride ion for the reduction.
Studies were also carried out to determine the optimum conditions for reduction. An excess of 2-4 equivalents of HCOONH4 was found to be ideal. The rate of transfer hydrogenation decreased substantially when only one equivalent of hydrogen donor was used. On the other hand, a large excess of HCOONH4 produced only a marginal increase in the rate of reaction compared to that observed when 2-4 equivalents were used. A large excess of catalyst improved the rate of transfer hydrogenation. We observed the optimal ratio of catalyst to substrate to be 2:1. Larger amounts of catalyst resulted in only minor improvement. However, the rate of transfer hydrogenation was significantly slower when a smaller amount of catalyst was used. A control experiment was carried out using oximes with ammonium formate or ammonium chloride, but without zinc dust, and the starting material was recovered in almost quantitatively. This clearly indicates that zinc catalyses the reaction. In anticipating metal/alcohol reduction, experiments were performed in absence of hydrogen donor by refluxing substrate with methanol and zinc dust for 4-5 hours. Even after a longer period the starting material was isolated in quantitative yield, which clearly indicates the requirement of ammonium formate or ammonium chloride for the reduction. Here methanol serves as a solvent.
Table 1
Transfer hydrogenation of oximes to amines catalysed by commercial
zinc dust using ammonium formate or ammonium chloride
| 1/2 | R1 | R2 | HCOONH4 |
NH4Cl |
bp (�C) |
|||
Time |
Yielda |
Time |
Yielda |
Found | Lit. |
|||
a | Ph | H | 2 min |
92% |
145 min |
88% |
182-184 |
18516 |
b | Me | H | 3 min |
62%b |
125 min |
60%b |
165d |
16516 |
c | Me | Me | 3 min |
70%b |
114 min |
67%b |
151d |
15016 |
d | Ph | Me | 4 min |
95% |
162 min |
90% |
184 |
184-18617 |
e | Ph | Ph | 5 min |
91% |
215 min |
88% |
295-296 |
29516 |
f |
p-OH-Ph-CH2 | Me | 3 min |
94% |
175 min |
85% |
123d |
125-12617 |
g |
p-MeO-Ph | H | 3 min |
92% |
160 min |
91% |
236 |
236-23716 |
h |
3,4,5-(MeO)3Ph | H | 5 min |
90% |
195 min |
81% |
122 |
12116 |
i |
p-Cl-Ph | H | 3 min |
94% |
152 min |
87% |
212-214 |
21516 |
j | C2H5 | CO2H | 4 min |
86% |
143 min |
83% |
302d |
30417 |
k | Ph | CN | 6 min |
81%c |
180 min |
74%c |
166d |
164-16516 |
l |
e | e | 4 min |
91% |
165 min |
83% |
100 |
10116 |
m |
f | f | 8 min |
91% |
195 min |
86% |
157-58 |
158-16016 |
n |
g | g | 35 min |
84% |
340 min |
71% |
135 |
13416 |
o |
h | h | 2 min |
92%c |
45 min |
89%c |
266-268d |
265-27012 |
- Isolated yields are based on single experiment and were not optimised.
- Low yield is due to low bp of the product (isolated as picrate derivative).
- Isolated as hydrochloride salt.
- Melting point.
- 1l 4-Nitrobenzaldehyde oxime, 2l 4-Aminobenzylamine
- 1m Succinaldehyde dioxime, 2m 1,4-Diaminobutane
- 1n Cyclohexanone oxime, 2n Cyclohexylamine
- 1o 4-Nitrocinnamic acid, 2o 4-Aminocinnamic acid
In conclusion, the reduction of oximes can be accomplished in a short time with zinc dust instead of expensive catalyst like palladium13-14. The yields are virtually quantitative and analytically pure. This zinc-catalysed procedure provides a very efficient, selective, inexpensive, rapid and is a general methodology for reduction of oximes to amines. Further investigations of other useful applications related to cleavage of peptides from resin support in solid phase peptide synthesis are in progress.
Experimental
The oximes were either commercially available or prepared from the corresponding carbonyl compound by standard methods. In cases where the oxime was obtained as an E/Z-mixture, no attempts were made to separate such mixtures.
Reduction of oximes to amines, general procedure:
To a solution of the substrate (5 mmole) in methanol (10 ml) was added ammonium formate (10-20 mmole) [or ammonium chloride (10-20 mmol)] and zinc dust (10 mmol). The mixture was stirred under reflux. After the completion of the reaction (monitored by TLC), the reaction mixture was filtered through celite. The filtrate was evaporated under vacuum and the residue was taken into chloroform or ether, washed twice with 80% saturated brine solution and finally with water. The organic layer was dried over anhydrous sodium sulphate and evaporation of the organic layer was followed by purification either by preparative TLC, or by column chromatography, to yield the desired product.