Abstract
α-Arylated ketones were obtained in moderate to good yields by one-step electroreductive coupling of α-chloroketones and arylhalides in DMF and in the presence of a Al- or Zn-sacrificial anode and a catalytic amount of a nickel complex.
1-Aryl-2-propanones are versatile intermediates for the synthesis of pharmaceuticals, agrochemicals, and fragrances. Two general approaches have been used to arylate ketones at the α-carbon1: i) the arylation of either a ketone enolate or equivalent from arylhalides or arylazosulfides via a SRN1 reaction, or a α-haloketone from arylboranes or arylcopper; ii) the acylation of benzylic organometallics. The reactions described so far do not however appear to be of wide scope, and are often multistep processes.
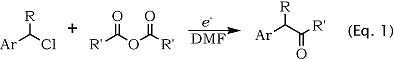
We already described2 a one-step electrochemical version of acylation reaction by direct electroreductive coupling of benzylic chlorides and acylanhydrides in a undivided cell and using a magnesium sacrificial anode and a stainless-steel cathode (Eq. 1).
This method however did not prove to be of very wide scope. We then turned to the arylation-type reaction, taking also into consideration that variously substituted arylhalides are more commonly available and easier to handle than benzylic halides.
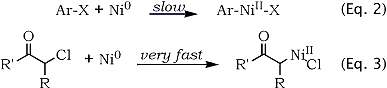
It is well known that arylnickel compounds can be coupled with various electrophiles such as CO2 or carbonyl groups3. The usual chemical routes to the arylnickel compounds however require the tedious preparation of Ni0 complexes such as Ni(cod)2. But there is an interesting alternative, already used for other syntheses4,5, which is the in situ electroreductive generation of Ni0 complexes in the presence of both the aromatic halide and the electrophile. The main advantages of the method are the use of an easily prepared Ni(II)bpyn complex in catalytic amounts and the easy control of the overall reaction. The feasibility of the reaction however requires that the electrophile does not react with Ni0 since the aromatic halide can not act as electrophile towards an alkyl nickel intermediate. Since for the reaction to occur, it has to go through the formation of an arylnickel intermediate, we anticipated that this relatively slow oxydative addition (Eq. 2) could only occur in the presence of quite a low concentration of any α-chloroketone which all react very rapidly with Ni0. This was easily obtained by slowly adding the ketone during the electrolysis. In addition, running the reactions at 80°C allowed us to perform the synthesis of arylacetones from arylbromides, while reactions involving aryliodides could be carried out at room temperature. We thus succeeded in preparing the compounds reported in Table 1 by coupling of aryl halides with chloroacetone (Eq. 4, R = H).
Table 1
Nickel catalysed electroreductive
coupling of chloroacetone and ArX
ArX (10 mmol) | AcCH2Cl (mmol) |
Product Yielda |
||||||||
Iodobenzene |
29 | 1 54% |
||||||||
4-Iodoanisole | 22 | 2 65% |
||||||||
3-Iodoanisole | 28 | 3 56% |
||||||||
Methyl 2-Iodobenzoate | 39 | 4 51%b |
||||||||
α-Iodonaphtalene | 39 | 5 23% |
||||||||
Bromobenzene | 35 | 1 62% |
||||||||
3-Bromo- benzotrifluoride | 27 | 6 79% |
||||||||
4-Fluoro- bromobenzene | 35 | 7 56% |
||||||||
4-Bromoanisole | 35 | 2 43% |
||||||||
5-bromo- 1,3-benzodioxole | 35 | 8 34% |
||||||||
2,5-Dimethoxy- bromobenzene | 35 | 9 34% |
||||||||
| ||||||||||

Table 2
Nickel catalysed electroreductive coupling
of α-chloroketones with aryl halides.
ArX (10 mmol) |
α-chloro- ketone |
mmol |
Product Yielda |
4-Iodoanisole | Phenacyl Chloride |
25 |
10 52% |
4-Bromoanisole | 3-Chloro- 2-butanone |
26 |
11 53% |
3-Bromo- benzotrifluoride | Phenacyl Chloride | 13 | 12 63% | 3-Chloro- 2-butanone | 12 | 13 70% |
p-Fluoro- benzonitrile | Phenacyl Chloride |
15 | 14 40% |
3-Chloro- 2-butanone |
31 |
15 43% |
|
4-Bromo- benzonitrile | 3-Chloro- 2-butanone |
13 |
16 70% |
3-Bromopyridine | 3-Chloro- 2-butanone |
22 |
17 45% |
a. Isolated yields, based on initial ArX.
Similarly, 3-chlorobutanone-2 and α-chloroacetophenone were cross-coupled efficiently with aromatic halides using the same procedure (Table 2).

In a preliminary study we also found that such a slow addition of the α-chlorocarbonyl reagent can allow to improve the coupling of α-chloroesters with aromatic halides (Eq. 4, R = H, CH3) which is not very efficient in many cases (especially with aryl bromides) when the two reagents are mixed in the cell at the beginning of the reaction6.
In conclusion, this nickel-catalyzed electrochemical coupling of aromatic halides with α-halocarbonyl compounds affords a valuable alternative to the methods of α-arylation of carbonyl compounds mentioned above.
| No. | Ketone |
|---|---|
1 | 1-Phenyl-2-propanone |
2 | 1-(4-Methoxyphenyl)-2-propanone |
3 | 1-(3-Methoxyphenyl)-2-propanone |
4 | Benzoic acid, 2-(2-oxopropyl)-methyl ester |
5 | 1-(1-Naphthyl)-2-propanone |
6 | 1-(3-[Trifluoromethyl]phenyl)-2-propanone |
7 | 1-(4-Fluorophenyl)-2-propanone |
8 | 2-propanone, 1-(1-3-benzodioxol-5-yl) |
9 | 1-(2-5-Dimethoxyphenyl)-2-propanone |
10 | 2-(4-Methoxyphenyl)-1-phenylethanone |
11 | 3-(4-Methoxyphenyl)-2-butanone |
12 | 2-(3-Trifluoromethylphenyl)-1-phenylethanone |
13 | 3-(3-Trifluoromethylphenyl)-2-butanone |
14 | 2-(4-Fluorophenyl)-1-phenylethanone |
15 | 3-(4-Fluorophenyl)-2-butanone |
16 | Benzonitrile, 4-(1-methyl-2-oxopropyl) |
17 | 3-(3-pyridyl)-2-butanone |
Experimental
Freshly distilled DMF (40 ml), Bu4NBF4 (0.6 mmol), NiBr2bpy (1 mmol), the aromatic halides (10 mmol) were introduced into a one-compartment cell fitted with Al rod/nickel-sponge or Zn rod/carbon fiber as the anode/cathode set of electrodes (cathode area ~20 cm2). The α-chloroketone was added constantly to the solution via a syringe pump at a rate of 0.5 ml/h and the electricity was supplied at constant current intensity of 0.25 A (nickel cathode) or 0.2 A (carbon cathode). Reactions involving aryl iodides were run at room temperature and those with bromides at 80°C. ArX was consumed after 4000 to 5000 coulombs were passed, which amounts around 1.5 Faraday per mole of the chloroketone. The reactions were then quenched with 4 N HCl and extracted with diethylether. Pure products were isolated by silica-gel column chromatography eluted with 90:10 or 85:15 pentane/diethylether.
All prepared compounds are known and were characterized on the basis of the agreement of their NMR data with literature data.