Abstract
Novel preparative methods for arylacetone and arylacetonitrile are described. Friedel-Crafts reactions of aromatic compounds with α-chloro-α-(methylthio)acetone (4) and α-chloro-α-(methylthio)acetonitrile (7) in the presence of Lewis acid afforded α-(methylthio)arylacetone (5) and α-(methylthio)arylacetonitrile (8), respectively. Compounds (5) and (8) we converted into the corresponding arylacetone (6) and arylacetonitrile (9) by reduction with zinc dust in acetic acid.

In the preceding paper,1 we showed that the Friedel-Crafts reaction of aromatic compounds with ethyl α-chloro-α-(methylthio)- acetate (1) provided an excellent method for synthesizing arylacetic esters (3) through reductive desulfurization of the reaction product (2). In the present paper the method is applied to syntheses of arylacetone (6) and arylacetonitrile (9), in which α-chloro-α-(methylthio)acetone (4) and α-chloro-α-(methylthio)acetonitrile (7) are employed as electrophiles in place of 1.2
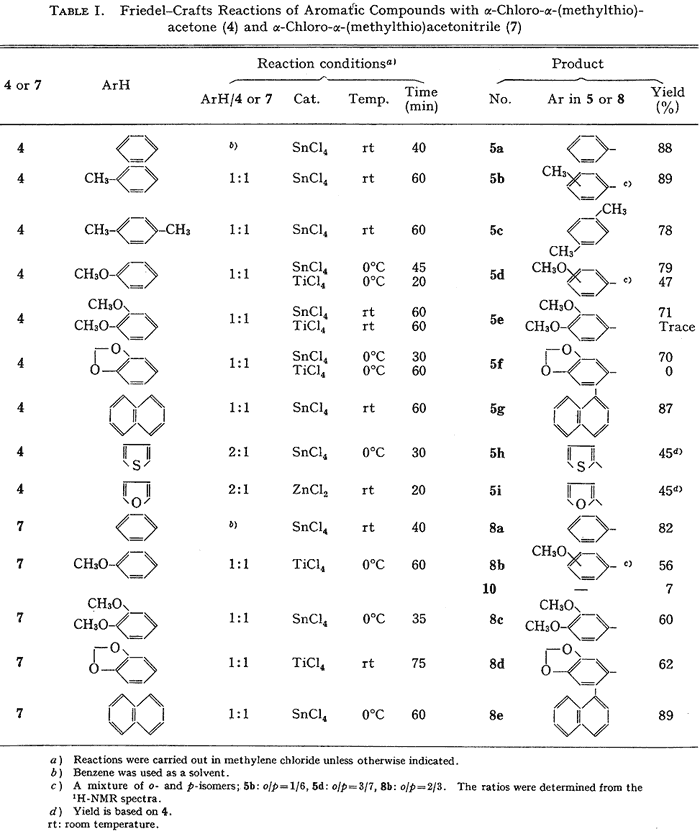
The previous study on Friedel-Crafts reaction with 11, revealed that the reaction requires one equivalent of Lewis acid, that no polyalkylated product is formed in the reaction, and that the order of activity of Lewis acids is SnCl4 = AlCl3 > TiCl4 >> ZnCl2. On the basis of this information, the Friedel-Crafts reaction of aromatic compounds with 4 was established. Thus, treatment of equimolar amounts of dimethoxybenzene and 4 in methylene chloride (CH2Cl2) with one equivalent of SnCl4 at room temperature gave α-(3,4-dimethoxyphenyl)-α-(methylthio)acetone (5e) in 71% yield. The results of the reactions of other arenes with 4 are summarized in Table I. The reactions generally take place smoothly in the presence of SnCl4 and give the adducts (5) in satisfactory yields.
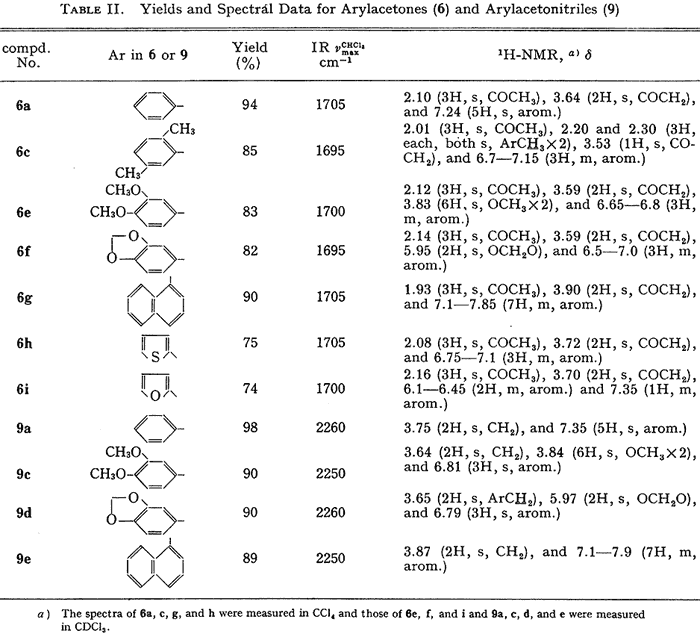
Desulfurization of 5 could easily be accomplished by treatment with zinc dust in hot acetic acid. The yields of arylacetones (6) are given in Table II.
Many synthetic methods for arylacetone have so far been reported in the literature. The following methods are representative: 1) thermal condensation of arylacetic acid and acetic acid in the presence of thorium dioxide at 430-450°C3, 2) Darzens condensation of arylaldehyde with α-bromopropionic ester followed by hydrolysis and decarboxylation of the resultant glycidic ester;4 and 3) photochemical condensation of arylbromide and acetone enolate ion in liquid ammonia.5 The present sequence of reactions can be performed under rather mild conditions, gives arylacetone in good yield, and is of preparative value.
Friedel–Crafts reaction of aromatic compounds with 7 was similarly carried out. Treatment of equimolar amounts of dimethoxybenzene and 7 in CH2Cl2 with one equivalent of SnCl4 at room temperature gave α-(3,4-dimethoxyphenyl)-α-(methylthio)acetonitrile (8c) in 60% yield. The results of the reactions of other arenes with 7 are summarized in Table I. In the reaction of anisole, the α,α-diarylacetonitrile (10) was formed as a byproduct in 7% yield with TiCl4.6
The adducts (8) were easily converted to the corresponding arylacetonitriles (9) in high yields by reduction with zinc dust in acetic acid (see Table II).
Of the several methods available for the preparation of arylacetonitrile, that most frequently employed is based upon reaction of arylmethylhalide with cyanide ion.7 The present method is useful as an alternative synthesis of arylacetonitrile without the use of cyanide ion.
Table I.
Friedel-Crafts Reactions of Aromatics with
α-Chloro-α-(methylthio)acetone (4) and
α-Chloro-α-(methylthio)acetonitrile (7)
{kind=link}
|
RCl
|
ArH |
Cat.
|
Temp
(°C) Time (min) |
Product & Yield | |||
|
4
| Benzene |
SnCl4
|
rt
|
40
|
5a
|
88%
|
|
| Toluene |
1:1
|
SnCl4
|
rt
|
60
|
5bc
|
89%
|
|
| p-Xylene |
1:1
|
SnCl4
|
rt
|
60
|
5c
|
78%
|
|
| Anisole |
1:1
|
SnCl4
|
0
|
45
|
5dc
|
79%
|
|
| Anisole |
1:1
|
TiCl4
|
0
|
20
|
5dc
|
47%
|
|
| Veratrole |
1:1
|
SnCl4
|
rt
|
60
|
5e
|
71%
|
|
| Veratrole |
1:1
|
TiCl4
|
rt
|
60
|
5e
|
Trace
| |
| 1,3-Benzo- dioxole |
1:1
|
SnCl4
|
0
|
30
|
5f
|
70%
|
|
| 1,3-Benzo- dioxole |
1:1
|
TiCl4
|
0
|
60
|
5f
|
0%
|
|
| Naphthalene |
1:1
|
SnCl4
|
rt
|
60
|
5g
|
87%
|
|
| Thiophene |
2:1
|
SnCl4
|
0
|
30
|
5h
|
45%d
| |
| Furan |
2:1
|
ZnCl2
|
rt
|
20
|
5i
|
45%d
| |
|
7
| Benzene |
SnCl4
|
rt
|
40
|
8a
|
82%
| |
| Anisole |
1:1
|
TiCl4
|
0
|
60
|
8bc
|
56%
|
|
| Veratrole |
1:1
|
SnCl4
|
0
|
35
|
8c
|
60%
|
|
| 1,3-Benzo- dioxole |
1:1
|
TiCl4
|
rt
|
75
|
8d
|
62%
|
|
| Naphtalene |
1:1
|
SnCl4
|
0
|
60
|
8e
|
89%
|
|
a) Reactions carried out in DCM unless indicated
b) Benzene solvent
c) Mixture of o-/p-isomers
d) Yield based on 4 used in the rxn rt: room temp
Table II.
Arylacetone (6) and
Arylacetonitrile (9) yields
{kind=link}
|
No.
|
Ar in 6 and 9 |
Yield
|
|
6a
|
Phenylacetone |
94%
|
|
6c
|
2,5-Dimethyl- phenylacetone |
85%
|
|
6e
|
3,4-Dimethoxy- phenylacetone |
83%
|
|
6f
|
3,4-Methylendioxy- phenylacetone |
82%
|
|
6g
|
1-(1-naphthyl)acetone |
90%
|
|
6h
|
1-Thien-2-ylacetone |
75%
|
|
6i
|
1-(2-furyl)acetone |
74%
|
|
9a
|
Phenylacetonitrile |
98%
|
|
9c
|
3,4-Dimethoxy- phenylacetonitrile |
90%
|
|
9d
|
3,4-Methylendioxy- phenylacetontrile |
90%
|
|
9e
|
1-naphthylacetonitrile |
89%
|
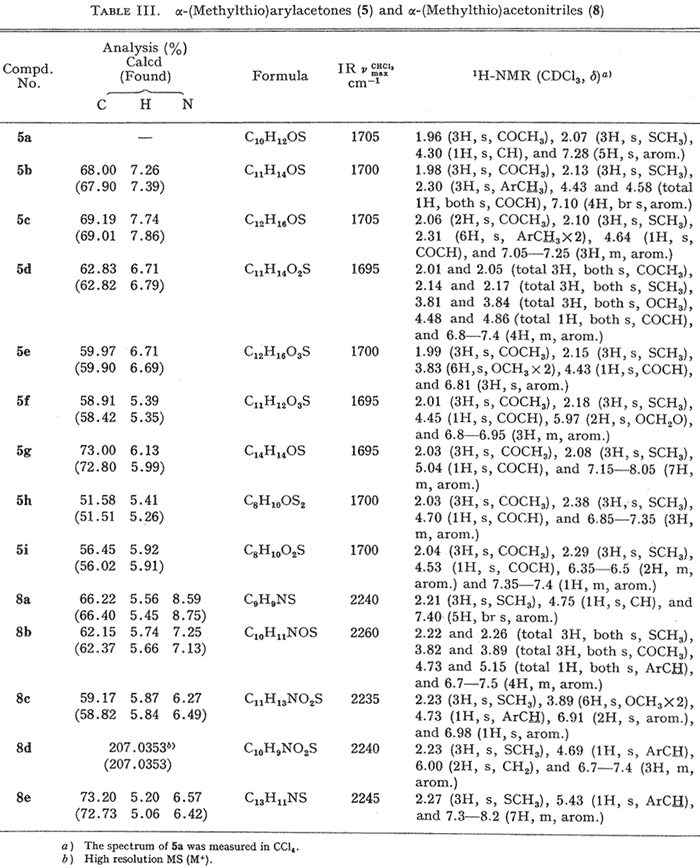
Table III.
α-(Methylthio)arylacetones (5) and
α-(Methylthio)acetonitriles (8)
{kind=link}
Experimental8
α-Chloro-α-(methylthio)acetone (4)
This compound was prepared according to the procedure described by Böhme.9 N-Chlorosuccinimide (13.9 g, 0.104 mol) was added to a stirred solution of α-(methylthio)acetone10 (10.4 g, 0.1 mol) in carbon tetrachloride (150 ml) in small portions at 0°C and stirring was continued at room temperature for 3 h. The precipitated succinimide was filtered off and the solvent was removed in vacuo. The residual oil was distilled to give 4 (8.82 g, 64%), bp 77.5°C (16 mmHg), lit.9 76-77°C (15 mmHg).
α-(Methylthio)phenylacetone (5a)
SnCl4 (1.03 g, 3.96 mmol) was added to a stirred solution of 4 (549 mg, 3.96 mmol) in benzene (9 ml) at 0°C, and stirring was continued at room temperature for 40 min. The reaction was quenched by the addition of water, and the mixture was extracted with benzene and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed on silica gel using benzene an eluent to give 5a (630 mg, 88%), which was identified by comparison of its spectroscopic data with those reported.11 The data are given in Table III.
α-(Methylthio)arylacetones (5b, c, d, e, f, g, h, and i); General Procedure
SnCl4 or ZnCl2 (for furan) (1.78- 1.89 mmol) was added to a stirred solution of 4 (1.78-1.89 mmol) and an aromatic compound [1.78-1.89 mmol except for the case of the reaction of thiophene or furan (2 eq)] in CH2Cl2 (10-15 ml) at 0°C, and stirring was continued under the conditions described in Table I. The reaction was quenched by the addition of water, then the mixture was extracted with CH2Cl2, and the extract was dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed on silica gel using benzene as an eluent to give 5b, c, d, e, f, g, h, or i oil. The yields and physical data are listed in Table I and III.
Arylacetones (6a, c, e, f, g, h, and i): General Procedure
Zinc dust (1 g) was added to a solution of 5a, c, e, f, g, h, or i (200-300 mg) in acetic acid (2-3 ml), and the resultant mixture was heated with vigorous stirring at 100°C for 1 h, then cooled. Water (20 ml) and CH2Cl2 (30 ml) were added, and the inorganic materials were filtered off. The organic layer was separated and the aqueous layer was further extracted with CH2Cl2). The combined organic layer was dried (MgSO4) and the solvent was evaporated off. The residue was chromatographed on silica gel using benzene as an eluent to give 6a, c, e, f, h, or i as an oil. The yields and physical data are listed in Table II. The compounds (6a),12 (6c)12, (6g),5 (6h),13 and (6i)14 were identified by comparison of their 1H-NMR spectra with those reported. The compound (6e) was identified by comparison of its IR and 1H-NMR spectra with those of a commercial sample. The compound (6f) was characterized as the 2,4-dinitrophenylhydrazone, mp 143-144°C (from ethanol).
α-Chloro-α-(methylthio)acetonitrile (7)
This compound was prepared according to the procedure described by Böhme.9 N-Chlorosuccinimide (11.07 g, 0.083 mol) was added to a stirred solution of α-(methylthio)acetonitrile15 (7.2 g, 0.083 mol) in carbon tetrachloride (45ml) in small portions at 0°C and stirring was continued at room temperature for 4.5 h. The precipitated succinimide was filtered off and the solvent was removed in vacuo. The residual oil was distilled to give 7 (5.52 g, 55%), by 67-69°C (12-14 mmHg), lit.9 65.5°C (12 mmHg).
α-(Methylthio)phenylacetonitrile (8a)
SnCl4 (1.09 g, 4.18 mmol) was added to a stirred solution of 7 (508 mg, 4.18 mmol) in benzene (2 ml) at 0°C, and stirring was continued at room temperature for 40 min. Work-up as described for the preparation of 5a gave 8a (560 mg, 82%), whose physical data are given in Table III.
α-(2- and 4-Methoxyphenyl)-α-(methylthio)acetonitriles (8b) and α,α-bis(4-Methoxyphenyl)- acetonitrile (10)
TiCl4 (412 mg, 2.17 mmol) was added to a stirred solution of anisole (234 mg, 2.17 mmol) and 7 (263 mg, 2.17 mmol) in CH2Cl2 (15 ml) at 0°C, and stirring was continued at the same temperature for 1h. The reaction was quenched by the addition of water, then the mixture was extracted with CH2Cl2, and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed on silica gel using benzene as an eluent to give 8b (235 mg, 56%) as an oil, whose physical data are given in Table III. Further elution with the same solvent gave 10 (38.3 mg, 7% based on 7), mp 153-154°C (from n-hexane).
α-(Methylthio)arylacetonitriles (8c, d, and e) General Procedure
SnCl4 or TiCl4 (for 1,2-methylenedioxybenzene) (3 mmol) was added to a stirred solution of 7 (3 mmol) and an aromatic compound (3 mmol) in CH2Cl2 (15 ml) at 0°C, and stirring was continued under the conditions described in Table I. The reaction was quenched by the addition of water, then the mixture was extracted with CH2Cl2, and dried (MgSO4). The solvent was removed in vacuo and the residue was chromatographed on silica gel using benzene as an eluent to give 8c, d, or e as an oil. The yields and physical data are listed in Table I and III.
Arylacetonitriles (9a, c, d, and e); General Procedure
Zinc dust (1 g) was added to a solution of 8a, c, d, or e (100-200 mg) in acetic acid (2-3 ml), and the mixture was heated with vigorous stirring at 100°C for 1 h, then cooled. Work-up as described for the preparation of 6 gave 9a, c, d, or e, which was identified by comparison of its physical data with those of a commercial sample. Yields and spectral data are given in Table III.