Abstract
An improved procedure to accomplish the O-phosphorylation of 4-hydroxy-N,N-dimethyl- tryptamine (Psilocin, 5) is reported that utilizes reaction between the O-lithium salt of psilocybin and tetra-O-benzylpyrophosphate. The O-benzyl groups were removed by catalytic hydrogenation over palladium on carbon to afford N,N-dimethyl-4- phosphoryloxytryptamine (Psilocybin, 1). In view of difficulties encountered in the preparation of 1, it is suggested that 4-acetoxy-N,N-dimethyltryptamine (2) may be a useful alternative for pharmacological studies. The latter was obtained following catalytic O-debenzylation of 4-benzyloxy-N,N-dimethyltryptamine in the presence of acetic anhydride and sodium acetate.
Recently, several laboratories have initiated clinical studies of hallucinogenic (psychedelic) agents1-3. This renewed interest suggests that there may be some demand for investigational substances that are suitably pure for human use that can be prepared in a relatively economical fashion. Hallucinogens are not commercially available in large quantities or in purities suitable for human studies, and research will likely be carried out only with drugs produced by custom synthesis. Of the various drugs that might be of interest for this work, most of them, including mescaline, LSD, DMT, and various substituted amphetamines are synthesized relatively easily. Indeed, many hallucinogens are routinely manufactured in clandestine laboratories.
By contrast, the synthesis of psilocybin, N,N-dimethyl-4-phosphoryloxytryptamine (1), is more challenging. Nevertheless, psilocybin has pharmacological features that make it attractive for clinical research, including a relatively short duration of action. The increasing worldwide popularity of psilocybin-containing mushrooms as recreational drugs also points to the need for more research with psilocybin. We re-examined the synthesis of psilocybin reported by Hofmann and coworkers4. Although their approach still remains useful, there were several weak points that could be addressed to improve the yields and purities of the final compound.

The overall synthetic route is shown in Scheme 1. The most troublesome step is the last, the phosphorylation of psilocin. In the original synthesis by Hofmann et al.4 the phosphorylation step was accomplished using O,O-dibenzylphosphorylchloride, an unstable reagent that was used without purification as a solution in carbon tetrachloride. Furthermore, the final yield of psilocybin was less than 20%. In view of the overall difficulty in preparing this material and its precursors, such a low yield in the last step was deemed unacceptable.
The present synthesis employs a phosphorylation step using tetrabenzylpyrophosphate, a stable, crystalline reagent. The phosphorylation step was complicated by the previously unreported extremely labile nature of the O,O-dibenzyl ester of psilocybin. Hydrolytic cleavage of one of the O-benzyl groups occurred rapidly in the presence of water, at room temperature, and neutral pH. The purification of the resulting zwitterionic material was much more complicated than for the basic O,O-dibenzyl material.
Scheme 1

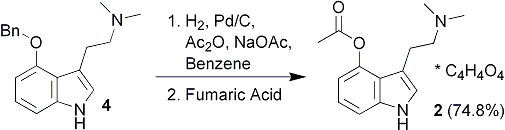
Illustrated in Scheme 2 is the facile preparation of 4-acetoxy-DMT52. This O-acetyl prodrug of psilocin is much more easily prepared than psilocybin, and may offer an economical alternative for clinicians wishing to study the psychopharmacology of psilocin. This material is readily crystallized as the fumarate salt, and is considerably more stable than psilocin itself. It would seem to be an ideal prodrug to replace psilocybin in future clinical studies, since psilocin is the principal metabolite of psilocybin.6
Scheme 2
The classical Speeter and Anthony synthesis of tryptamines from indoles served as the precedent for this work.7 The key reaction of oxalyl chloride with 4-benzyloxyindole was, however, sluggish. Similarly, the reduction of the 4-substituted glyoxalylamide 3 was much slower than for indoles without substitution at this position. TLC was used to monitor the complete disappearance of starting material and intermediate reduction products. The O-benzyl group was then readily removed by catalytic hydrogenolysis to afford 4-hydroxy-N,N-dimethyltryptamine (psilocin; 5).
After experimentation with a variety of phosphorylating agents, it was finally decided that tetrabenzylpyrophosphate (TBPP) was the most suitable reagent.8 This crystalline and stable material is commercially available (Aldrich), but can also be synthesized readily on a multigram scale.
The most convenient base for the phosphorylation step proved to be butyllithium. Generation of the lithium salt of psilocin in THF at -70 °C, followed by addition of 1.1 equivalents of TBPP, led to the O,O-dibenzyl ester of psilocybin, with the generation of one equivalent of lithium O,O-dibenzylphosphate that must ultimately be removed. While ordinarily removal of the lithium salt would not be problematic, washing the organic reaction mixture with water led to unexpected and rapid hydrolysis of one of the O-benzyl groups. Judicious exclusion of traces of water allowed the isolation of O,O-dibenzyl ester that was nearly free of 6. The O,O-dibenzyl intermediate proved to be so sensitive to water, however, that it was more practical to use an aqueous workup, allow hydrolysis to occur, and isolate a product that was largely the zwitterionic O-monobenzylphosphate 6.
Catalytic hydrogenolysis of the crude O-benzyl ester led to the formation of psilocybin (1). The procedure was complicated by small amounts of phosphoric acid generated from residual dibenzylphosphoric acid carried from the previous step into the hydrogenolysis reaction. This highly acidic material leads to discoloration of the product and prevents satisfactory crystallization. The problem was solved through the use of anion exchange resin to titrate the phosphoric acid. The reported pH of a solution of psilocybin in 50% aqueous ethanol is 5.2.9 Anion exchange resin (OH- form) was added in portions, with vigorous and extended stirring, to the filtered reaction solution until the pH of the solution was 5.2.9 When this pH was reached, the resin was removed by filtration and the filtrate was concentrated under vacuum. The crude product was then recrystallized from a small amount of methanol, and a large volume of isopropanol, followed by storage in the freezer. Psilocybin (1) crystallized as long colorless needles.
As a potential replacement for (1), 4-Acetoxy-N,N-dimethyltryptamine fumarate (2) was conveniently prepared by shaking under hydrogen a mixture of 4, acetic anhydride, and sodium acetate in benzene with Pd/C in a Parr low pressure hydrogenation apparatus. Following uptake of the required amount of hydrogen corresponding to O-debenzylation, the catalyst and insoluble salts were removed by filtration. One molar equivalent of fumaric acid was added to the filtrate, and the solution was concentrated to dryness under vacuum. The resulting solid was recrystallized to afford white crystals of the desired product. This material was stable when stored in the cold, but slowly darkened on storage for several months at ambient temperature.
Experimental
4-Benzyloxyindol-3-yl-N,N-dimethylglyoxylamide (3)
A solution of 4-benzyloxyindole (17.5 g, 0.078 mol) (Biosynth) in anhyd Et2O (500 mL) was mechanically stirred in a 1 L, 3 necked flask and cooled in an ice-salt bath to an internal temperature of 0°C. Oxalyl chloride (20.3 g, 0.16 moles) was added dropwise at a rate that maintained an internal temperature between 0-5°C. Stirring was continued for 3 h at a temperature between 5-10°C with a gentle argon sparge to remove evolved HCl. The argon sparge was replaced by a gas inlet tube and a dry ice/acetone condenser. Anhyd dimethylamine was then bubbled into the reaction with cooling and vigorous stirring until a pH (determined by moist pH paper) between 9 and 11 was achieved. At this time, the orange color of the initial solution had been mostly discharged, and the reaction had the appearance of a slightly off-white slurry with a few flecks of yellow unreacted starting material. CH2Cl2 (20 mL) was added to assist solubilization of the unreacted material and the reaction was stirred for an additional 6 h to yield finally an offwhite slurry. Et2O (150 mL) was added, and the mixture was cooled to 10°C. The white solids were collected by suction filtration on filter paper in a Buchner funnel and then were suspended in distilled H2O (250 mL) and stirred for 1 h to remove dimethylamine hydrochloride. The slurry was filtered, and the collected solids were washed on the filter with distilled H2O (3 x 75 mL) and hexane (75 mL) and dried overnight in a vacuum oven. The dried product weighed 18.3 g. The organic filtrates and washes were combined and the solvent was removed by rotary evaporation. The residue was dissolved in CH2Cl2 (100 mL) and the organic solution was washed with distilled H2O (2 x 50 mL) and brine (2 x 50 mL). After drying (MgSO4) the volume was reduced by rotary evaporation. The concentrated residual solution was subjected to flash chromatography over silica gel, first eluting with CH2Cl2 to recover unreacted indole (1.3 g, 7.4%), followed by elution with 10% MeOH in CH2Cl2 to recover 3.3 g of 3. The latter was combined with the initial product to provide a total weight of 21.6 g (85.9%). The crude product was recrystallized from MeOH/EtOAc to give 19.5 g (77%) of 3 with mp 152-155 °C (Lit.4 mp 146-150 °C).
4-Benzyloxy-N,N-dimethyltryptamine (4)
A slurry of LiAlH4 (8.90 g, 0.234 mol) in anhyd THF (100 mL) was prepared in a 2 L, 3-neck flask, previously dried with a heat gun under an argon purge. The flask was fitted with a reflux condenser, mechanical stirrer, and addition funnel. Anhyd dioxane (200 mL) was added, and the mixture was heated to 60°C on an oil bath. 4-benzyloxyindol-3-yl-N,N-dimethylglyoxylamide (3) (14.5 g, 0.045 moles) was dissolved in a mixture of dioxane (250 mL) and THF (150 mL) and, with rapid stirring, this solution was added dropwise over 1 h. The oil bath temperature was held at 70°C for 4 h, followed by vigorous reflux overnight (16 h) at an oil bath temperature of 95°C. Thin layer chromatographic analysis (9:1 CH2Cl2/MeOH silica plates) showed nearly complete reduction. The reaction was heated at reflux for an additional 4 h and then cooled to 20 °C. A solution of distilled H2O (27 mL) in THF (100 mL) was added dropwise, resulting in a gray flocculent precipitate. Et2O (250 mL) was added to assist breakup of the complex and improve filtration. This slurry was stirred for 1 h and the mixture was then filtered with a Buchner funnel. The filter cake was washed on the filter with warm Et2O (2 x 250 mL) and was broken up, transferred back into the reaction flask and vigorously stirred with additional hot Et2O (500 mL). This slurry was filtered, and the cake was washed on the filter with Et2O (150 mL) and hexane (2 x 150 mL). All of the organic filtrates were combined and dried (MgSO4). After the drying agent was removed by filtration, the filtrate was concentrated under vacuum at 40°C and dried under high vacuum at 0.01 mmHg, leading to crystallization of the residue as a white waxy solid. Recrystallization from EtOAc yielded 12.57 g, (94.8%) of 4 with mp 124-126 °C (lit.4 mp 125-126 °C).
4-Hydroxy-N,N-dimethyltryptamine (Psilocin, 5)
A solution of 4 (4.0 g, 0.0135 moles) in 95% EtOH (250 mL) was added to 1.5 g Pd/C (10% w/w) in a 500 mL Parr low pressure hydrogenation bottle. The mixture was shaken under 60 psig of H2 pressure for 2 h. The catalyst was removed by vacuum filtration through Celite and was washed on the filter with EtOH (3 x 50 mL). The filtrate was concentrated by rotary evaporation. The clear residual oil was placed under high vacuum and induced to crystallize by seeding. The white crystalline powder (2.68 g, 97.0%) was used in the next step without further purification.
4-O-Monobenzylphosphoryloxy-N,N-dimethyltryptamine (6)
A solution of 0.45 g (2.2 mmol) of psilocin (5) and 0.073 g (0.73 mmol) of diisopropylamine in anhyd THF (50 mL) was magnetically stirred in a 100 mL 3-necked flask and was cooled to -78 °C in a dry ice-acetone bath. A 2.5 M solution (1.14 mL, 2.85 mmol) of n-BuLi in hexane was added dropwise using a syringe. After complete addition, the reaction was stirred for 3 min and tetrabenzylpyrophosphate8 (1.50 g, 2.8 mmol) was added all at once. The dry ice-acetone bath was replaced by an ice-salt bath, and stirring was continued for 1.5 h. TLC (9:1 CHCl3-MeOH alumina plates) showed complete disappearance of starting material. The reaction was quenched by addition of sat. NH4Cl (30 mL). The biphasic solution was rapidly transferred to a separatory funnel, and the aqueous layer was separated and washed with EtOAc (2 x 20 mL). The organic layers were combined and washed with brine (25 mL), followed by drying (anhyd MgSO4). The solution was then concentrated to a clear residue using rotary evaporation. This residue (1.12 g) was shown by thin layer chromatography and NMR analysis to be a mixture of O,O-dibenzylpsilocybin, O-monobenzylpsilocybin (6), and a small amount of dibenzyl phosphoric acid.
N,N-Dimethyl-4-phosphoryloxytryptamine (Psilocybin; 1)
In a 250 mL Parr hydrogenation bottle was placed 1.0 g of 10% Pd/C catalyst followed by anhyd MeOH (50 mL). The dibenzyl/monobenzylphosphoryloxy-N,N-dimethyltryptamine (1.12 g) prepared in the previous step was added and the mixture was shaken under 60 psig hydrogen pressure for 3 h, at which time hydrogen uptake had ceased. The hydrogenation bottle was removed from the apparatus and the catalyst was removed by filtration through a pad of Celite 545 on a Buchner funnel. The pH of the clear solution was measured at 3.7 using a pH meter. Amberlite IRA-400 anion exchange resin (OH- form) (0.75 g) was added in 3 portions to the well-stirred methanolic solution to raise the pH to 5.3.9 The resin was removed by vacuum filtration and the resulting clear filtrate was concentrated to dryness by rotary evaporation. The residue was dissolved in a minimum amount of hot MeOH, and hot isopropanol was added to the cloud point. An additional drop of MeOH produced a clear solution. Upon storage in a -20°C freezer the product slowly crystallized as white needles 0.294 g (46.9%, from psilocin). This product was dried under high vacuum to produce solvent-free psilocybin, which had mp 212-213°C (lit.5 mp 210-212 °C).
4-Acetoxy-N,N-dimethyltryptamine5 fumarate (2)
In a 250 mL Parr hydrogenation bottle was placed 0.25 g of 10% palladium on charcoal followed by anhyd sodium acetate (1.50 g, 18 mmol). Benzene (50 mL) was added, followed by acetic anhydride (5 mL, 5.41 g, 5.32 mmol), and 4 (0.50 g, 1.7 mmol). The mixture was shaken under 60 psig of hydrogen for 4 h. After the uptake of hydrogen had ceased the hydrogenation bottle was removed from the apparatus, the mixture was diluted with THF (25 mL), and the catalyst was removed by filtration through a pad of Celite 545. The catalyst was washed repeatedly with isopropanol (3 x 50 mL). The washings and mother liquor were collected separately because of unreacted Ac2O in the filtrate. The mother liquor was concentrated under vacuum to about one half the original volume, then toluene (50 mL) was added. The solution was again concentrated by rotary evaporation. The isopropanol washes were combined with the residue and also concentrated. The residue was then dissolved in anhyd MeOH (50 mL). Fumaric acid (0.198 g, 1.7 mmol) was dissolved in MeOH (10 mL) and added to the stirred methanolic solution of the residue. After stirring for 10 minutes, toluene (50 mL) was added and the solution was concentrated to dryness by rotary evaporation. Absolute EtOH was added to the residue and a white precipitate of 2 fumarate (0.290 g, 0.8 mmol) formed and was collected by filtration. The filtrate was evaporated and the residue was dissolved in a minimum amount of MeOH. EtOAc was added and clear crystals began to form. After storing the solution in a freezer at -10°C, 0.170 g of additional product was collected for a total yield of 0.460 g (74.8%); mp 172-173°C.