Chapter 6 in Handbook of Psychopharmacology, Volume 11: Stimulants
(Edited by Leslie L. Iversen Susan D. Iversen and Solomon H. Snyder), Plenum Press, New York 1978
HTML by Rhodium
The field of psychopharmacology has its sources in a number of different medical disciplines involved with the function of the brain. In the area of pharmacology, a major contribution has been made by the study of those drugs that are collectively known as psychotropic agents. The term "psychotropic" is used to describe a drug that turns or changes the mind, following quite literally from the Greek stem ψνχη) meaning soul, mind, or understanding, and τρεπειν, to turn. A sizable proportion of our present pharmacopoea contains medicines designed to play a role in the changing or modification of a person's mood or mental state. A classification of these materials into families defined by the nature of the mental state change evoked is a useful way of defining the subdivision known as "psychotomimetic drugs." An historically interesting procedure was established by Lewin (1924), who characterized these drugs in five subdivisions according to the nature of their action.
a. Excitantia. This group is composed of drugs that, when used in modest amounts, produce a central stimulation, an increase in alertness, and a relief from fatigue. Many of these drugs have come from plant sources and have been intimately spun into our social behavior patterns, Coffee, tea, and tobacco are so generally accepted that they are in fact no longer considered drugs. Similarly, other societies have also brought botanical agents, such as kola, betel nut, and khat, into their daily behavior patterns. The most important of the component alkaloids of many of these plants are caffeine and ephedrine, and both have found extensive medical application. An extremely effective local anesthetic, cocaine, has been broadly used for its central stimulation effects.
In current medical practice most of the drugs employed for achieving this form of psychotropic action are synthetic in origin, and are based largely upon the chemical structure of amphetamine, The many variations and evolutions of this paradigm drug have been discussed elsewhere in this volume, as have their various clinical uses.
b. Inebriantia. The ubiquitous social maneuver of "getting drunk" is a form of psychopharmacological toxicity that is widely recognized and generally acceptable, The best known drug in this classification is, of course, ethanol, This material has been incorporated, in one form or another, into virtually every culture in the history of man. The syndrome of intoxication, the generalized loss of inhibition and intellectual control, is easily recognized and quite consistent in appearance. Drinking has become a social ritual and many people have lost sight of the fact that alcohol is an effective, although low potency, psychotropic drug, Virtually all of the drugs that have found use as central anesthetics or as hypnotics can provoke, in the early stages of intoxication, a disinhibition similar to that of alcohol, and many have been used to provide this form of "social" intoxication, Ether (by itself, or mixed with alcohol), nitrous oxide, chloroform, trilene (trichloroethylene), and the hydrocarbons such as benzene and hexane are among the many volatile chemicals that have been employed as intoxicants, Many of the clinically effective sedatives have been used, with appropriate attention to dosage, in this manner.
c. Hypnotica. As suggested above, there is a very close connection between excitement and hypnosis. Most of the drugs that, at clinically effective levels, effect hypnosis (a sleeping state from which there can be arousal but little recall) have proven to be disinhibitors and intoxicants at subclinical levels. The barbiturates, the quinazolones (such as methaqualone and mecloqualone), the carbamate paralytic tranquillizers (such as meprobamate), and the benzodiazepines (as chlordiazepoxide and diazepam) can all show stimulation and an alcohollike intoxication. These responses are usually merely minor side-effects, but can become major symptoms during the early stages of intoxication (or during chronic light medication) when there is social reinforcement to forestall or divert overt hypnosis.
A major pharmacological class of drugs, the anticholinergics, should be classified in this area, Although commonly thought of as psychotomimetic, these compounds have been employed clinically as anesthetics. There is no question but that there are vivid psychotropic aspects of the intoxication produced by plants such as belladonna and jimson weed, or of chemicals such as ditran and phencyclidine, but the amnesiac properties of these drugs sets them apart from the larger family of psychotomimetics. Their use leads to a loss of contact with reality and a sensory isolation, but the substantially complete lack of recall of the events that occur during intoxication places these substances closer to the anesthetic class. However, there is at no point a clinical loss of consciousness, although they have been used medically in close association with anesthetics.
d. Euphorica. Lewin chose this name for a group embracing the compounds he felt to be mental sedatives rather than soporific physical sedatives, His major entry, opium and the alkaloids found in opium, still represents the chemical point of departure for most of the drugs that are today classified in this division. The sensory analgesics in this area are characterized not by an amplification of sensory awareness, nor by a confusion of it, but by a replacement of it with a euphoric insulation wherein problems and worries are of less importance, The best known drug in this classification, one that has played a major role in the structuring of both legislation and medical practice, is morphine. Morphine, in addition to being a central analgesic, was broadly championed in the late nineteenth century as a safe alternative for alcohol in medical alcoholism cases. When its capability to produce a physical dependence itself was appreciated, its diacetate, heroin, was employed as a nonaddictive cure for morphine addiction. It was used in this role for more than a decade, until its own dependence liability was understood.
A second role that has been played by morphine in this euphorica classification has been to serve as the structural prototype for an incredible cascade of analgesic drugs, a large number of which have maintained the psychotropic pharmacology of heroin.
e. Phantastica. Phantastica or hallucinatoria was the name given by Lewin to encompass many drugs that he held as "thaumaturgic agents - which evoke sense - illusions in a great variety of forms." At the time of his writing, only a few plant sources had been recognized, and their actions could be accounted for by a handful of chemicals. Peyote, the Banisteriopsis snuffs, marijuana, and the Amanita mushrooms were known, but only a few of these had been analyzed with sufficient interest or care to provide drugs that satisfactorily reproduced the actions of the natural intoxicant, Today, many additional plant sources have been studied, and the art of the chemist and the pharmacologist has swelled the list of examples to well in excess of 100. A comparison of the structures, and an attempt to correlate their potency and qualitative actions with these structures for the most broadly represented groups of these, the phenethylamines and alpha-methylphenethylamines, will be the purpose of this chapter.
The contrived word "psychotomimetic" finds its origin in the combination of "psychoto-" from psychosis, and "-mimetic" from the Greek μιμητικος meaning "in imitation of." There has been an inordinate amount of confusion and conflict in the search for agreement upon a name for this class of compounds. The difficulties stem from an inadequacy of describers for the nature of the induced intoxication. Lewin's term phantastica stemmed from his holding that the effects of these drugs were "mysterious and incomprehensible." An incapability of adequately naming them is evident from phrases such as "the chief character of the visions (this, in reference to mescaline) is their indescribableness" (Ellis, 1898), and "we enter [upon taking these compounds] a world beyond language, so it is hardly surprising that they may be difficult to name" (Kl�ver, 1928).
A number of the names essayed to define this field of central intoxicants within the medical community have grown directly out of the symptomatology of intoxication. An occasionally encountered symptom of drug effect is a distortion of some sensory modality with the perception of a synthetic feature that upon objective analysis appears to be undocumentable. This event, commonly called a hallucination, has provided the term "hallucinogen," or literally, a material that gives rise to a hallucination. Hellpach (1941) proposed the term "eidetika" to put emphasis upon the particular nature of the hallucination evoked. With special emphasis upon the disruption of the central nervous system, Delay (1959) suggested the name "psychodysleptic" to place these drugs in direct comparison with those which depressed the mood (psycholeptics) and those which stimulated the mood (psychoanaleptics). The parallels of the drug-induced intoxication and mental illness have given rise to the term "psychotica" (Haase, 1966) and the above-mentioned, broadly used term "psychotomimetic" (Lehmann, 1958, 1959). These latter terms are aimed toward the research branch of the medical community, since they support the concept of the pharmacological generation of a "model psychosis." All of these terms are faulted in that the properties that have inspired them are not invariably seen or agreed upon, and at best they represent only one aspect of a very complex intoxication.
In the literary community, an entirely different family of names has been employed, with origins from Homer to Huxley. One of the most widely accepted has been the one suggested by Osmond in 1956, "psychedelic." Choosing the prefix "psyche-," to represent the mind or soul (he eschewed the proper spelling "psycho-" due to its resemblance to the word psychosis and the connotation of mental illness), a number of syntheses were proposed:
| psychephoric | (mind-moving) | |
| psychehormic | (mind-rousing) | |
| psycheplastic | (mind-molding) | |
| psychezymic | (mind-fermenting) | |
| psycherhexic | (mind-bursting-forth) | |
| psychelytic | (mind-releasing) | |
| psychedelic | (mind-manifesting) |
These terms all give particular emphasis to the reports and beliefs that these drugs can enrich the mind and can be explored as a positive tool in mental research, rather than giving continuous reinforcement to the negative features of intoxication (disruption, psychosis, and δνς or "bad" in dysleptic). The last of these terms, psychedelic, caught the fancy of the popular press, and with the unfortunate lay promotion of these drugs during the late 1960s became the ubiquitous adjective for many aspects of the drug culture. Its use as a describing term for these drugs now carries with it the subtle implication of approval of uncontrolled drug use for entertainment purposes, if not the active promotion of drug-intoxication philosophy. For this reason the word is seldom encountered in the scientific literature.
Many other names have been created to represent this class of drugs; each of them reflects some classical point of reference or some particular aspect of the induced intoxication. Some additional examples that have appeared on occasion in the literature are:
| delirients | pharmakons | |
| delusionogens | psychosomimetics | |
| dysleptics | psychotaraxics | |
| misperceptinogens | psychoticants | |
| mysticomimetics | psychotogens | |
| phanerothymes | psychogens | |
| phantasticants | schizogens |
and agents that are eidetic, hypnogogic, hypnopompic, psychoactive, psychotoxic, and consciousness- expanding.
This thesaurus is continuously expanding, as new emphases are desired or new predilections are apparent. For this chapter, we shall limit our vocabulary to the single, and admittedly inadequate word, psychotomimetic.
If there is confusion in choosing a term to describe the class of drugs that we shall call the psychotomimetics, then there is chaos in agreeing upon a description of their effects. To a large measure, their peripheral effects are minor and not deserving of attention. It is the area of the state of awareness modification, the changes in interpretation of one's environment, and the property of a catalytic unfettering of imagination that have commanded the psychiatrist's attention and the public's fancy.
Hofmann (1959) has described this as follows:
The psychotomimetics produce profound and acute changes in the sphere of experience, in the perception of reality. changes even in space and time and the consciousness of self. Phenomena of depersonalization may also occur. Retaining full consciousness, the subject experiences a kind of dream world, which in many respects seems to be more real than the customary normal world. Objects and colours, which generally become more brilliant, lose their symbolic character, they stand detached and assume an increased significance, having, as it were, their own more intense existence.
At the intellectual level, the experience has been analyzed as containing contributions from a number of components (Pahnke and Richards, 1966): a psychotic aspect, a truly dysphoric element that gives weight to the term psychotomimetic; a psychodynamic contribution, usually shown as the availability to the subject of subconscious and preconscious material; a cognitive component, characterized by astonishingly lucid thought; an esthetic value, which is realized by an increased perceptual ability in all sense modalities; and a transcendental or mystical quality. This aspect of the intoxication syndrome has been described as an experience wherein "one replaces a real world with an alternate real world which is equally real and yet different" (Shulgin, 1970a). The many facets of the psychotomimetic intoxication have assumed as many names, and each needs extensive definition and illustration before being safely employed. One can find reference to subjective phenomena such as positive (and negative) hallucinations, including pseudohallucinations; visualizations, including illusions, dread-states, eidetic images (with or without reality character, memory images (including projected images, afterimages, and pseudomemory images), reperceptions; d�j� vu phenomena; depersonalization (or derealization) phenomena; Sinnenged�chtnis; and no on. The fine structure of these psychological nuances is discussed in detail in this volume by Hollister in Chapter 8.
The complex generalized symptomatology ascribed to the use of psychotomimetic drugs is an amalgamation of thousands of studies involving dozens of different drugs. When one drug at a time is considered, an emphasis on one or another aspect of this syndrome is generally encountered. This would be of a peculiarity felt to be characteristic of the specific drug in question. But this very fractionation of symptoms of intoxication can give rise to a new type of difficulty in making drug-to-drug comparisons. An observer may be attentive to some expected facet of intoxication, and thus measure the effectiveness of a drug by whether this particular clue is or is not elicited.
It is most important that there be maintained a close communication between the experimental subject and the observer. Such rapport will allow an intellectual probing that can alert the observer to the nature and the depth of intoxication. There is also the need to minimize the varieties of expression that the experiment may take; in practice this is most easily realized by restricting the dosage of drug to be administered to those which may be expected to produce a modest or threshold stage of intoxication. If the subject is also the observer, then this communication problem must be met by exposure to a reproducible environment, equipped with familiar clues, which will potentially provoke recognized sensory distortions, and with some form of record-keeping that will permit facile recall of the details of the experience and yet not distort the experience itself.
An additional complication that must be considered in the description of the effects of a psychotomimetic drug is the unpredictable variability that may exist from person to person. This variability can exist both in the qualitative as well as the quantitative response to a drug, and the two are intimately interwoven. Some agreement must be made as to the nature of the drug response that shall be considered adequate before any value can be placed upon its potency.
False positives are not uncommon, wherein a subject, due to a combination of expectations, anxiety, and some form of self-hypnosis, can believe that he is experiencing a real effect following exposure to what eventually proves to be an ineffective level, or a placebo. In an effort to minimize these errors, the technique of employing an "active placebo" is occasionally reported. All of the psychotomimetic drugs, in addition to eliciting central effects, have minor, secondary indicators of activity as side-effects. There may be indications of mydriasis, light-headedness from blood-pressure changes, paresthesia, nausea, perhaps just a generalized feeling of discomforture. With a person who is familiar with these prodromal signs, there may be the subconscious synthesis of indicators of central disruption leading to a false positive response. This complication might be controlled by employing as a control an agent that mimics these aspects of toxicity but that is not psychotomimetic. False negative responses are rare.
The most desirable way of circumventing the problems inherent in individual variability between experimental subjects is to evaluate the effects of several drugs within a single subject. This allows, as far as possible, a consistent background of past experience and of sensory sensitivity, permitting the design of a true control component in clinical experimentation.
With the acceptance of two drugs being qualitatively similar in their action, the remaining problem in a structure�activity relationship is to compare their relative potencies. As has been mentioned above, and in detail in the chapter by Hollister, at drug dosage levels that produce a complete intoxication the nature of this intoxication can be most variable. The dosage requirements to produce such a degree of disruption will strongly reflect the specific parameter that is used to measure this disruption. A frequently used stratagem to minimize this variability is to exploit the observation that at threshold levels of intoxication there is seen quite a consistent constellation of clues. These are sufficiently easily recognized to distinguish active drug from placebo, but not sufficiently elaborate to define the drug state itself, the state that would be characteristic of the specific drug being evaluated. This technique is excellently illustrated by the studies of Abramson and Rolo (1967) in the quantitative comparisons of psilocybin, LSD, methysergide, and several homologous lysergic acid amides. An excellent consistency was shown among his experimental subjects in detecting threshold action at drug dosage levels that were not sufficiently high to permit the identity of the drug to be deduced. Additional advantages to this approach to quantitative analysis are that the clinical environment is obviously more easily managed, and that there is a minimum of psychological turmoil on the part of the experimental subjects.
The property of tolerance induction is a well-recognized pharmacological fact. This introduces yet another degree of uncertainty in the quantitative ranking of psychotomimetic drugs. To the extent that an experimental protocol calls for multiple drug exposures within the same individual, there is the possibility of the response to a later drug exposure being affected by an earlier drug experience. Fortunately there is little difficulty encountered from this source. Studies that have explored the development of tolerance, either from one drug to its own effects, or from one drug to a chemically related drug through chronic low-level exposure, have shown that there is frequently the quick development of an appreciable tolerance, but that this tolerance is as quickly lost. The usual prudence of adequate spacing of experimental challenges seems to be quite enough to minimize errors from this source.
A major paradox confronts the experimenter in the decision whether to use subjects who are experienced and are familiar with the effects of the psychotomimetic drugs, or whether to use subjects who are pharmacologically naive. A principal advantage with the experienced subject is the familiarity with the extraordinary symptoms that can develop along with the drug's induction of intoxication. Recognition of unnatural sensory input permits a relatively unemotional record to be made of the events taking place. The conviction that the experiment is transient and reversible minimizes the anxiety component of the syndrome. Mention has been made earlier concerning the appearance of false positive reactions from the misinterpretation of the physical distress clues that can precede a psychotomimetic experience, but to a drug-sophisticated subject these dues themselves, with an active compound, can alert him to the possibility of active threshold levels.
On the other hand, such a population can also contain people who, for one motive or another, may tend to exaggerate or even falsify their reports of a drug's effects. The use of prisoners as subjects is clearly compromised by some tacit understanding that pleasing the observer might produce fringe rewards. The use of subjects who are enthusiasts of one or another feature of psychotomimetic drug intoxication may lead to distorted reports motivated by the desire to have further opportunity to participate in such experiments. These criticisms are difficult to answer.
The use of naive subjects presents different problems. There can be complicated ethical questions that are not encountered with drug experiments that lead to more prosaic central nervous system changes. The small but real possibility exists of effecting long-term changes, or changes that are slow to reverse themselves. The risks of precipitating some long-lived psychotic state can be largely circumvented by the appropriate screening of the experimental volunteer. However, there are numerous examples on record in which the intensity of the psychotomimetic experience and the insight and self-evaluation that it can inspire have led to changes in personal priorities and even in social behavior patterns. These risks from both long-term effects and from possible immediate adverse reactions have been summarized by Cohen (1960). In general, most research clinicians in these areas have preferred to avoid the responsibilities inherent in these latter examples and have restricted themselves to subjects with some past drug experience.
A number of different conventions can be found in the scientific literature for the designation of the dosages employed in clinical studies. Most frequently one finds the weight of the dosage administered, although the physical form (free base, salt of some acid) is usually omitted. In an attempt to parallel the conventions in pharmacological research, this is often presented as the dosage of drug per weight unit of the subject (i.e., 5 mg/kg), but if the weight of the subject is not reported, the total dose can only be approximated. The usefulness or appropriateness of this per weight presentation is questioned by many researchers, especially with drugs that are effective on neural systems. The neural complexity of a person appears to be quite independent of his body weight. A further unnecessary refinement that has recently become quite popular is the expression of a drug's weight in terms of moles. This form precludes the ambiguities of salt-form mentioned above, but of course it cannot be applied to botanical extracts, to mixtures, or to other preparations of unknown molecular weight.
For reasons of consistency all dosage values presented in this chapter are converted (if necessary) to weight of drug administered, as specified, to an experimental subject of about 75 kg.
One of the greatest disappointments to the scientists involved in research in the area of the psychotomimetic drugs, has been the failure to find a satisfactory screening process or assay procedure that will duplicate the human psychopharmacological intoxication syndrome in animal models. Some of the better studied psychotomimetics are effective agonists or antagonists in biological in vitro systems, and these have been explored as potential screening tools. The close biochemical relationship between LSD and serotonin has been exploited to offer proposed mechanisms of action of the former drug, but efforts to extrapolate these relationships to structural analogs of LSD have led to disappointing correlations with human effectiveness. A constant stumbling block, as an example, is 2-bromo-LSD (BOL), which is as active an antiserotonin agent as LSD, but which is substantially without psychotomimetic activity in man. Within small chemical families, there has been promising correlation between in vitro titration and in vivo effectiveness; the anticholinergic activity of a family of glycolate esters (Abood, 1968). the sympathetic stimulation such as mydriasis and hyperthermia in a family of amide variations of lysergic acid (Cerletti, 1959); serotonin agonist potency in a homologous series related to DOM (Shulgin and Dyer, 1975). Most assays are faulted by showing valid correlations over only a small and closely related group of compounds, although a recent study by Aldous et al. (1974) suggests that rabbit hyperthermia might serve to bridge the gap between LSD, DOM, and mescaline (which covers three orders of magnitude difference in human dose requirements).
A number of promising assays have been explored based on body response or on behavioral changes induced in the intact animal. The hyperthermia observations have already been mentioned. Horita and Dille (1954) observed that rabbits were very responsive to small quantities of LSD (0.5 µg/kg), showing a rise in body temperature apparently of central origins. A reasonably good parallel in rabbits between this hyperthermia response and the known "excitatory syndrome" of LSD was extended throughout the family of lysergic acid amides and paralleled quite closely the reported human psychopharmacological potency (Hofmann, 1960). This work was extended by Jacob and his co-workers (Jacob et al., 1962; Jacob and Lafille, 1963), and Brimblecombe and co-workers (Brimblecombe it al., 1964; Brimblecombe, 1967) found that the parallel could be extended to the tryptamines. Aldous et al. (1974) have shown its applicability to the phenethylamine-like psychotomimetics discussed in this chapter, and it is felt that this is probably the best animal test at present for estimating psychotomimetic potency.
Several behavioral approaches have been studied. The use of unrestrained or untrained animals was the basis of Hall's open-field test (Hall, 1934) in which animal activity patterns (rearing, preening, defecation, ranging) were found to be influenced to a degree proportionate to a drug's potency in man. Lipman et al. (1963) have applied this test to a series of piperidinoglycolates, and Brimblecombe (1963, 1967) to a number of tryptamines. Behavioral tests in other animals [in mice, a head-twitch assay (Coyne and Pickering, 1967) and interference with nest-building (Schneider and Chenoweth, 1970); in cats (Brimblecombe et al., 1964), and a sham-rage response syndrome (Benington et al., 1958); in monkeys (Hunt and Brimblecombe, 1967)] are usually restricted to small groups of closely related compounds. A behavioral response assay (the Bovet-Gatti drug profile) has been restructured to allow for the evaluation of psychotomimetic drugs (Smythies et al., 1969). A recent review (Brawley and Duffield, 1972) has analyzed the extensive literature concerning these correlates. A failing with most of these assays is the need to employ large doses of the drug. In most cases these are approaching the lethal dose, and are certainly well above the dose/weight equivalent employed in man. Several specific assays have been critically analyzed (Silva and Calil, 1975) and it has been shown that not all families of drugs are validly detected and certain CNS agents that are not psychotomimetic respond as if they were; in general it is concluded that they are of limited value.
In attempts to circumvent some of these biological model limitations and to explore possible molecular correlates of action, a number of research groups have evaluated the physical-chemical properties of the psychotomimetic drugs. A number of attempts have involved the actual calculation of molecular parameters. Interatomic separations within a molecule would influence intermolecular hydrogen bonding to other molecules or to potential sites of action (Smythies et al., 1970; Kelley and Adamson, 1973). Intramolecular conformations are possible that might allow one active psychotomimetic to resemble another (Snyder and Richelson, 1968). A number of groups have made energy calculations at various orbital sites within groups of known psychotomimetics (Snyder and Merrill, 1965; Rang and Green, 1970) but in the one case where such studies were directed to a very narrow chemical class (LSD homologs), Kumbar and Siva Sankar (1973) found a poor correlation with human potency.
A number of molecular properties have been studied that are amenable to experimental measurement. Studies of crystal lattice geometry of active compounds by Baker and co-workers (1973) and Chothia and Pauling (1969) have provided three-dimensional portraits of molecular conformation, but extrapolation to an in vivo solution environment leaves such results difficult to interpret. Three physical-chemical approaches have overcome this theoretical difficulty by employing solutions in their analyses. Sung and Parker (1972) have estimated that the sr-bonding potential of a number of known active psychotomimetics through a spectroscopic measurement of the strengths of the charge-transfer complexes formed with p-dinitrobenzene. An extensive partition coefficient study (Barfknecht et al., 1975) has shown a fair correlation between the values of 1-octanol:water partition (either measured or calculated) and human activity. They suggest that a partition of about 1400:1 (at pH 7.4) is optimal for members of the phenylisopropylamine family of psychotomimetics, with loss of potency with change in either direction. The native fluorescence of dilute solutions of several known psychotomimetics has been determined and correlated with human activity (Antun et al., 1971).
A logical outgrowth of this situation is the frequent incidence of self-experimentation. The quintessence of informed consent is to be found in a clinical study where the designer and author of the experimental protocol is also the experimental subject. Alles (1959) first used the term "double-conscious" to emphasize this resolution to the problems concerning this area of research. He explained: "Might as well call this [a] 'double-conscious' technique, because I not only made the compound, but I weighed it out, dissolved it in water, and knew that I took it at [a] particular time. I was entirely on my own resources in observing what was happening." The use of a restricted group of subjects, drawn largely from the research team itself, circumvents many of the problems discussed earlier associated with paid or otherwise rewarded volunteers.
An important feature in the clinical evaluation of psychotomimetic drugs is the influence of the immediate environment within which the experiment is conducted. This "setting" not only plays an important role in establishing the attitude and tone of the subject's interpretation of the events that are occurring, but it can constitute a rich source of sensory clues. A consistent source of such "catalysts" is especially desirable in the comparison of two different drugs in a single subject. The hospital environment with its white walls, institutional sounds and smells, and constant associations with illness and medical authority has on occasion contributed a psychotic note to the drug experiment. For these reasons, many researchers prefer to conduct their drug evaluations in more unconventional settings such as the outdoors or in a person's home, and with the availability of sensory stimuli that are to a subject's own liking. On the other hand, Elkes et al. (1955) have expressed concern for the occasionally severe or delayed response seen, and have urged the use of a hospital environment with trained personnel and emergency facilities. These variables can promote or suppress the expression of a psychotomimetic episode and can certainly contribute to the diverse statements of drug potencies that are found in the literature.
The legal problems associated with this area of research cannot be ignored. Many of the psychotomimetic drugs, for the very reasons that they are known drugs of abuse and have no accepted medical utility, are classified as Schedule I drugs in the Controlled Substances Act. Medical research with these requires the presentation of an exacting protocol to several government and academic agencies for approval. It is maintained that these complications were not intended to restrict research in these areas but were merely to maintain some control over drug abuse possibilities; but these requirements certainly have had a dampening effect on research. Most of the materials to be discussed in this chapter are not included in any drug legislation, for they have never been implicated in any social problem. Work with this broader group of compounds must still recognize the public health and pharmacy laws, but is not an immediate concern of the narcotics law agencies.
Much of the exploration and use of the psychotomimetic drugs has taken place not in the clinic under the control of attending research scientists but in the paramedical areas of social drug use and noninstitutional research. The sources and identities of the drugs that are used can only be poorly documented. Some are certainly stolen, or diverted, from legitimate channels, and as such are of potentially firm identity and purity, although there can be misrepresentation in distribution. Many, however, are privately or clandestinely prepared.
Some of these preparations are entirely for purposes of exploration motivated by personal curiosity. The ready availability of chemical laboratories in the academic and industrial world can provide the facilities, and the volume of production is usually small since the use of the drug is generally restricted to the synthesist and his immediate acquaintances. Many structural variations are explored by intellectual curiosity. The investigator may be a student who is following an unrelated line of study; he may be an amateur chemist who is professionally employed in an unrelated field; he may be a physician who feels that to reveal this form of research would bring criticism from his peers.
Many preparations are, however, manufactured in frankly illicit laboratories. These can be production operations, motivated by the economics of drug sales rather than by the subtleties of drug effects. Here, the encouragements for structural variation are largely intentional changes designed to circumvent the exacting letter of the law that defines a restricted drug, or to compensate for the unavailability of some preferred starting material.
On occasion samples of such preparations are obtained in amount and in consistency sufficient to allow identification of the drug present and its connection to examples of documented use. Also, there is a sizable body of legitimate scientific information concerning the activity or the lack of activity of potential psychotomimetic drugs that has never been published for the reasons mentioned. Where appropriate, and in those cases where the information is felt to be reliable, these observations will be included in this compilation.
The several score of known psychotomimetic drugs based on the phenethylamine substructure will be organized and presented in this section in a manner that emphasizes their chemical character. This represents the "S" of the SAR, or structure-activity relationship.
A structure-activity study is a comparison of the chemical structures of a class of compounds with one another, and a correlation of these differences with observed differences of drug potency and drug effect. The chemical structure or formula is one of the few incontestable factual properties known and usually forms the framework of all relationship presentations. One of the underlying reasons for most of the studies made with the psychotomimetic drugs is the desire to explore the relationship of their action to endogenous mental illness. Their structures, compared with the naturally occurring biochemicals and neurochemicals, may explain their action or may indicate unexpected biochemical transformations, which can be involved with spontaneously expressed psychological problems. At the first approximation, it is usually assumed that the more potent a compound is, the more closely its structure will resemble some natural feature that is responsible for mental aberration. This assumption is faulted for a number of reasons. Relative potencies of compounds will reflect many other properties than just their absolute potency at some hypothetical site of action. Relative differences of absorption rate will exist. Even within a closely related series of compounds, differences in biotransformation are to be expected; many of these aspects are considered in detail with these compounds by Castagnoli in Chapter 7. The compound may serve well or poorly as a substrate to some enzyme system that has no bearing whatsoever on its relationship to native biochemicals. The tissue distribution of a drug may reflect its physical properties more than its metabolic vulnerability or inertness.
As has been stated above, only one of the two major families of psychotomimetic drugs will be considered here in great detail; this is the phenethylamine group, with the logical extensions to the alpha-methyl homologs, the homologs related to the chemical structure of amphetamine. This family has been employed in the discussions on metabolism as the presentation basis for the many aspects of biotransformation presently known. It will be used here as the basis for illustrating the many nuances of dependency known between minor chemical change and resulting change in the nature of the psychological intoxication that results from these changes. The second large family of psychotomimetics are best generalized as the indoles. Although they will not be considered in detail, they should be briefly presented in summary, for reference purposes.
The principal subdivision of the indole psychotomimetic drugs contains the substituted tryptamines. Most of those which have been studied are unsubstituted on the aromatic ring. One principal structural variation has been the manipulation of the identity of the substituent on the basic tryptamine nitrogen. N,N-Dimethyltryptamine (DMT) is the prototypic example, and human studies are known for the N,N-diethyl, N,N-dipropyl, N,N-diisopropyl, N,N-di-(n)-butyl, and N-mono-(t)-butyl homologs. The compounds with short-chain substitutions are only active parenterally, but the branched-chain counterparts are orally active. The substitution of a hydroxyl group at the 4-position yields, with the simplest member DMT, the natural alkaloid psilocin. This base, and its naturally occurring phosphate ester psilocybin, are orally active tryptamines and have also served as the basis of a series of homologs. The addition of a methoxy group to the 5-position generally maintains the route of application requirements of the 5-H counterpart, but increases the potency by a factor of 5 to 10. Fuller details on the structure-activity relationships known about the tryptamine psychotomimetics may be obtained from recent reviews and compilations describing them (Brimblecombe and Pinder, 1975; Shulgin, 1976a).
The second of the three indole subdivisions of the psychotomimetic drugs is a small but potentially very important branch. This involves the tricyclic carbolines and is best represented by the natural base harmaline. These materials were first introduced into human psychopharmacological study by the South Indian native use of the plant extract Ayahuasca. Botanically, there is a continuing stream of new compounds being uncovered in this area of native drug use. A very close connection has been established between the carbolines and the tryptamines in plant biosynthesis. In human biochemistry, these connections are also becoming understood with the widely recognized facile formation of the pyridine ring from normally occurring indoles such as serotonin. For a review of the structures and the psychopharmacology of the known active carboline psychotomimetics, see Schultes and Hofmann (1973) and Naranjo (1967, 1973).
Most of the drugs that are known today have had their origins in the family of chemicals known as the alkaloids. These are basic, nitrogen-containing organic chemicals from the plant kingdom, and they represent a bewildering array of structural variations. A consistent theme found through most of the alkaloids is the separation of the nitrogen atom from an aromatic system, by two carbon atoms:
This relationship can be found in most of the known families of alkaloids and has been the mainstay of the thousands of synthetic drugs that have been based upon some alkaloid model. It may be complexly substituted, or convoluted by additional ring forms or three-dimensional bridging, but it is usually there to be found.
The simplest aromatic system encountered is the benzene ring, and one of the largest classes of psychotomimetic drugs are the substituted phenethylamines. 3,4,5-Trimethoxyphenethylamine (mescaline, 1) will be considered as a prototype chemical, and variations on its structure will be the basis of the classification of this section.
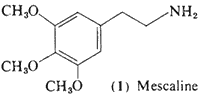
a. History. The story of mescaline (1) has its origins in the use of the cactus Lophophora williamsii, or Anhalonium lewinii. This dumpling cactus, known natively by the name peyote or peyotl, has been involved in North American Indian practices for centuries before Columbus, with its appearances in the funerary art of some 2000 years ago (Furst, 1972). The first description of peyote was made by Hernandez (1651), who called it peyote zacatecensis. His observations of its appearance, actions, and scarcity are beautifully concise, and have been quoted by Schultes (1972):
The root is nearly medium size, sending forth no branches or leaves above the ground, but with certain wooliness adhering to it... It appears to have a sweetish and moderately hot taste. Ground up and applied to painful joints, it is said to give relief... This root . causes those devouring it to foresee and predict things.. or to discern who has stolen from them some utensil or anything else, and other things of like nature... On which account, this root scarcely issues forth, as it it did not wish to harm those who discover a and eat it.
All written record of pre-Spanish culture was lost, and the only sources of the native rituals of its use are to be found in the more remote desert and mountain areas where there was sufficient cultural isolation to maintain some of the original history. For descriptions of the northward migration of peyote culture in the late nineteenth century, reference should be made to the writings of Slotkin (1956) and La Barre (1969) and the review of Marriott and Rachlin (1971).
Occurrence of Mescaline in Cacti
| Cactus | Locale | Reference |
| Lophophora williamsii | Texas, Chihuahua | Heffter (1898) |
| L. diffusa (Anhalonium williamsii) | Queretaro | Todd (1969) |
| Trichocherus pachanoi | Peru | Poisson (1960), Agurell (1969a) |
| T. bridgesii | Bolivia | Agurell (1969a) |
| T. macrogonus | S. America | Agurell (196th) |
| T. terscheckii | Argentina | Reti and Castrill�n (1951) |
| T. werdermannianus | S. America | Agurell (1969b) |
| T. cuzcoensis | Peru | Agurell et al. (1971) |
| T. fulvilanus | S. America | Agurell et al. (1971) |
| T. taquimbalensis | S. America | Agurell et al. (1971) |
| T. validus | S. America | Agurell et al. (1971) |
| Stetsonia coryne | Argentina | Agurell et al. (1971) |
| Pelecyphora asilliformis | San Luis Potosi | Neal et al. (1972) |
b. Cactus Sources. The best known botanical origin of mescaline is the aforementioned peyote cactus L. williamsii, which is found throughout the Rio Grand area of Texas and Northeastern Mexico, and well south into the state of Chihuahua. A related species, L. diffusa, occurs yet further south in the state of Queretaro (Bravo, 1967), but it has been reported to contain only traces of mescaline (Todd, 1969). Heffter (1898) reported analyses of A. lewinii and A. williamsii and found great variation in mescaline content. It is now felt that the L. diffusa species was actually at hand, and these chemotaxonomic problems have been largely resolved (Braun, 1975).
A large number of cacti have been considered to be psychoactive, and collectively they have been referred to as the "peyote complex." This is in part due to their physical resemblance to L. williamsii, and in part to the reputation of toxic effects associated with their use. Included are examples of the genera Ariocarpus, Astrophylum, Aztekium, Dolichothele, Obregonia, Pelecyphora, and Solisia. There has been a recent flurry of analytical research into the alkaloid content of these plants, and a large number of compounds have been reported that are either present in L. williamsii or can be biosynthetically related to them. Mescaline itself, however, has only been reported in the hatchet cactus Pelecyphora asilliformis (Neal et al., 1972).
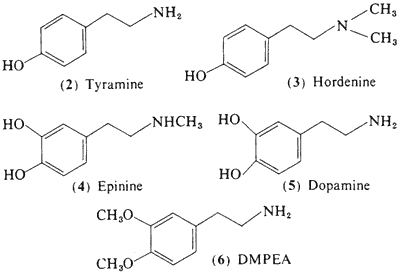
c. Congener Alkaloids. Brief mention should be made concerning the alkaloids that are found in company with mescaline in the peyote cactus. Many of these can be classed as less-complexly substituted phenethylamines. These are interesting in their probable roles as biosynthetic precursors of mescaline (Paul, 1973). A number of these are recognized pharmacologically active drugs: tyramine (2) (McLaughlin and Paul, 1966); hordenine (3) (McLaughlin and Paul, 1965); and epinine (4) (Lundstr�m, 1971a,b). Others are recognized biochemicals normally found in the body: dopamine (5) (Lundstr�m, 1971a) and 3,4-dimethoxyphenethylamine, DMPEA (6) (Lundstr�m and Agurell, 1968). The amounts present in peyote appear to be too small to contribute to the intoxication syndrome that follows plant ingestion.
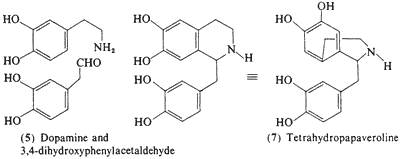
More interesting from the point of view of contribution to the psychopharmacological effects of the cactus peyote are the tetrahydroisoquinolines that are present. At the present time there are some 25 reported as being identified in the plant (see Kapatia and Fayez, 1973). The theoretical interest in several of these compounds comes from their possible in vivo formation by the closing of the amino group of a phenethylamine with some aliphatic aldehyde. This process has been suggested as a reasonable procedure for the generation of alkaloid-like compounds in mammalian species, through the interaction of the phenethylamines and the corresponding phenylacetaldehydes to generate tetrahydropapaveroline (7) and morphinelike compounds (Davis and Walsh, 1970), These structural manipulations have been discussed in the chapter on biotransformations.
In peyote, four of the tetrahydroisoquinoline alkaloids that accompany mescaline are known to produce some central activity in man, and may conceivably contribute to the psychopharmacology of the plant. These are all condensation products On effect) of a phenethylamine and acetaldehyde. Pellotine (peyotline, 8) is the major alkaloid reported by Heffter (1898) in the A, williamsii now believed to be L. diffusa (see above). Although there appears to be no reports of field work that describes the use of this latter species, its morphological similarity to active peyote would suggest that it might have played a role in Indian religious rites. The psychopharmacological action of pellotine in man is one of sedation rather than intoxication, however. At low levels (15-30 mg) there is a calming effect and the observation of the production on an uneventful sleep in patients with dosages of 50 mg subcutaneously (Jolly, 1896). Heffter (1898) reported that at levels of as much as 240 mg (total dose) there is a dizziness and a generalized tiredness but no indications of sensory distortions.
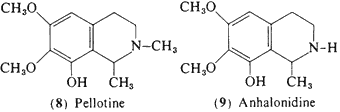
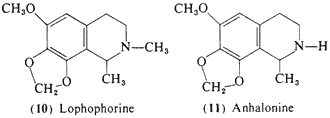
Anhalonidine (9) appears to produce a heavy-headedness and sedation similar to that of pellotine, but is about one fourth as potent, Dosages of between 100 and 250 mg produced a marked sedation, but again without any reported sensory changes, Lophophorine (10) in animal studies is the most toxic of the components of peyote, and in man an oral dose of 20 mg provoked vasodilation, an immediate headache, and a warm flushed feeling (Heffter, 1898). A 50-mg dose produced a marked slowing of the heart with a compensatory rise in blood pressure, but no suggestion of mescaline-like effects (Dixon, 1899), Anhalonine (11) has been evaluated in a single experiment with an oral dose of 100 mg and this led to an uneventful tiredness without any observed central effects of a sensory nature (Heffter, 1898).
d. Dose and Route. Mescaline is usually administered at a dosage level of between 300 and 500 mg, in the form of the sulfate salt (equivalent to 225-375 mg as the free base), Dosages at the higher limit are often administered in two portions, spaced about an hour apart, to lessen the abruptness of onset, and to minimize the nausea that is usually produced by the drug. The drug has been administered by a number of routes (oral, subcutaneous, intramuscular, and intravenous) without changes in the dose requirement for intoxication, although with the intravenous route the first effects are noted immediately (Hoch, 1951). With this route, the period of maximum intoxication occurs somewhat sooner than usual (1.5-2 hr following the drug's administration) but the overall time period is largely the same as that seen with other routes of administration.
e. Psychopharmacological Syndrome. Many books, reviews, and research reports have appeared with extensive detail of the mescaline-induced intoxication syndrome. Reference should be made to Beringer (1927) and Rouhier (1926) for a body of clinical detail that will outline the variability of the individual response to the drug. A generalized profile of effect has been constructed from a number of separate descriptions (Shulgin, 1973a):
At a nominally active dosage level of 350 mg (orally, as the sulfate) there is a generally predictable chronology of events. The first signs of change arc largely physical. At about a half hour following ingestion there is an onset of nausea. often accompanied with active vomiting. There is occasionally the development of diarrhea. A mild tachycardia and rise blood pressure is often seen during this initial phase. but this may be associated with anxiety and apprehension. The initial indication of sensory change is noted in about one hour. The development of central effects ends the "physical distress" phase of the intoxication and this "sensory" phase continues to develop vi a plateau of intensity during the next two to three hours. The physical changes noted during this period are minor. There is a cardiovascular quieting with the pulse rate and blood pressure dropping below their initial base levels, and a constant, extensive, but reactive, mydriasis, A gradual diminution of the central intoxication over the following few hours leads to a complete recovery, generally within twelve hours. There is consistently an excellent recall of the impressions and events that occurred during the experiment.
Whereas this time pattern and sequence of events is quite predictable from one person to another and from one occasion to another, the content and the direction taken by the subject's imagination as directed by his interpretive capacities are completely unpredictable and are unique to each experience.
Some sensory changes are regularly noted and can be expected to contribute to the overall impact of the drug's effects. There is a shimmering and intensification of the visual field, far more intense than what one might expect from the mydriasis-induced photophobia. There is an intensification of color perception, and extreme amplification of minor differences in both color and texture. Frequently observed is the generation of patterned imagery, sometimes in a grid structure, sometimes with undulating shapes, but usually with some color contribution. There is a benign empathy shown to both inanimate and living things, especially to small things.
There is still no satisfactory answer to the question of the action of mescaline being possibly different from the reported effects of the entire peyote cactus, Thus far, there have been no reports that have compared the chemical with the plant in a single study. The two substances are usually used in quite different ways: the chemical is administered as a single acute bolus, to an isolated individual, in a clinical setting that can provide overtones of social improperness. On the oither hand, the sacramental use of peyote involves consumption over an extended period of time, in a group situation, and is unquestioned as an accepted ritual. Some of these factors surely contribute to the reported differences in the induced intoxication state.
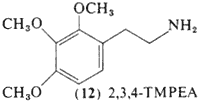
2,3,4-Trimethoxyphenethylamine (12, 2,3,4,-TMPEA) is a positional isomer of mescaline first prepared by Slotta and Heller in 1930. Direct pharmacological comparisons of the drug with mescaline in conditioned response assays showed the two to be essentially equivalent in terms of both dosage and nature of response (Winter, 1973) although it was much more rapidly oxidized in mouse brain homogenate preparations (Seiler and Demisch, 1971).
A single report exists concerning the activity of this isomer of mescaline in man, The following is translated from the report of Slotta and M�ller (1936):
We have discovered that the intoxicating action depends to a remarkable degree upon the position of the three methoxy groups. Mescaline, the 3,4,5-trimethoxy-beta-phenethylamine produces in the normal subject a much stronger overall intoxication than in the schizophrenic patient. whereas 2,3,4-trimethoxy-beta-phenethylamine has quite the opposite effect. It has little action in healthy individuals, being almost without intoxicating properties, but it is very potent in the schizophrenic. The metabolic conversion products of the "reciprocal" mescaline will be further studied as soon as the study of the metabolism of the proper mescaline is complete.
The dosage employed of the 2,3,4-trimethoxy isomer is not given, but the mescaline dosages were 400 mg and it can be presumed that a similar amount had been used in the "reciprocal" mescaline experiments, The promised further studies have not as yet appeared.
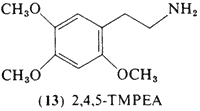
This positional isomer of mescaline, 2,4,5-trimethoxyphenethylamine (13, 2,4,5-TMPEA) was first synthesized by Jansen (1931) and compared directly with mescaline. Pharmacological studies in both frogs and cats led to the conclusion that the two compounds were qualitatively similar. In this study, unspecified amounts of 2,4,5-TMPEA
were administered (by injection) to the author, and finally a control test was carried out with an equal quantity of mescaline. The action of both these substances agreed only to a limited extent with the effects described for mescaline by, for example, Beringer (1927). It must be remembered, however, in this connection, that the quantities used by Beringer were larger than the doses administered in these experiments. Nevertheless it may be concluded, that the pharmacological action of beta-2,4,5-trimethoxyphenylethylamine agrees to a large extent to that of mescaline. However the new compound had more unpleasant secondary effects (nausea) and did not bring about the euphoric state caused by mescaline.
It is not possible to estimate from this quotation the potency of 2,4,5-TMPEA. The smallest dose unit reported by Beringer was 300 mg, so that lesser amounts than this were presumably employed in this experiment. A more recent study (Dittrich, 1971) found that an acute oral dosage of 300 mg was indistinguishable from placebo, with no report of any such "secondary" effects, Although not centrally active itself, 2,4,5-TMPEA appeared to potentiate the action of mescaline when employed as pretreatment 45 min prior to the administration of mescaline, Smythies et al. (1967a) had reported that 2,4,5-TMPEA did not appear to be hallucinogenic in rat model studies, in substantial agreement with the acute study of Dittrich. However, these animal studies suggested that 2,4,5-TMPEA should have an inhibitory action against mescaline (Smythies, 1967b) rather than the potentiating action found.
This compound is the trimethyl ether of the extremely interesting neurological poison 6-hydroxydopamine and one should expect to uncover valuable information by further exploring this amine in clinical studies.
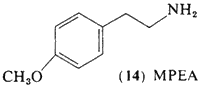
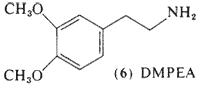
Interest has been directed toward the simpler methoxylated phenethylamines for a number of years, Ernst (1962, 1965) had observed that the lower homologs of mescaline, 4-methoxyphenethylamine (14, MPEA) and DMPEA (6) produced a mescaline-like catatonia in mice, a property that is absent in the corresponding phenols. Michaux and Verly (1963) reported that the mono-methoxy compound (14) was the most biologically active of these methoxylated phenethylamines, MPEA (14) as well as DMPEA (6) has been found as a component of human urine (Sen and McGeer, 1964).
Brown et al. (1968) have studied the effects of MPEA in man. Sixteen normal subjects were given MPEA at dose levels of approximately 400 mg orally, employing mescaline as a standard in the same subjects, and at the same dose. All of the subjects reacted as expected to the mescaline administration, and none of them showed any response whatsoever to MPEA.
3,4-Dimethoxyphenethylamine (6, DMPEA) has been the center of controversy for over a decade. The initial report of the occurrence of this compound in the urine of schizophrenic patients (Friedhoff and Van Winkle, 1962) has been confirmed by several independent investigators. The findings of Perry et al. (1964) that DMPEA was not present in the urine of schizophrenic patients has also been confirmed by several independent investigators. Dietary factors have been implicated (von Studnitz and Nyman, 1965), a cyclic nature of the appearance of DMPEA has been observed (Kalbhen and Braun, 1973), and the application of radioimmunoassay techniques has indicated that small, erratic levels may be present in all human subjects (Knoll and Wisser, 1976). This last analytical technique is extremely sensitive but appears to show some cross reactivity to normal urinary metabolites (Riceberg and Vunakis, 1975).
DMPEA has received additional attention because of its close chemical relationship with the known neurotransmitter dopamine (5). The processes of enzymatic O-methylation may give rise to DMPEA in the intact individual and this latter compound, with its resemblance to mescaline, is an attractive candidate for the role of an endogenous psychotogen.
This point has been evaluated by the direct measurement of DMPEA as a psychotomimetic in man. A series of studies in normals and in schizophrenic patients showed that oral dosages between 400 and 1000 mg were without either central or peripheral effects (Friedhoff and Hollister, 1966; Shulgin et al., 1966; Charalampous and Tansey, 1967; Hollister and Friedhoff, 1966; Brown et al., 1968). It was only at a remarkable 1500 mg that subtle changes in behavior were observed (Vojtechovsky and Krus, 1967), and these were compared to the stimulant effects of caffeine.
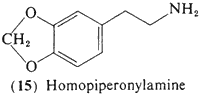
3,5-Methylenedioxyphenethylamine (15, homopiperonylamine) contains the methylenedioxy group that is characteristic of several of the alkaloids present in peyote, This compound, and the 5-methoxy analog homomyristylamine (18) are biosynthetically related to a large number of plant products. The three-carbon homologs (to be discussed later) of these two bases have been shown to be effective psychotomimetics in man and so with their close resemblance to the known peyote alkaloids prompted their evaluation as potential psychotomimetics. A single report exists concerning trials with homopiperonylamine (15) (Alles, 1959), and it was found to be without the slightest peripheral or central effects following two separate assays of 200 mg.
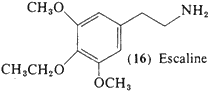
Literally hundreds of alkoxylated analogs and homologs have been synthesized, and many have been explored in biochemical and pharmacological studies. However, 3,5-dimethoxy-4-ethoxyphenethylamine (16, escaline) and the two following entries (17, 18) are the only analogs of mescaline, differing only in the identity of the substituent group on the ether oxygens, that have been assayed as psychotomimetics in man, Acute studies with (16) have shown it to he active orally in the range of 40-60 mg (Nichols and Shulgin, unpublished data), It differs from mescaline in that the onset of action is quicker (within the first hour) and there is no nausea noted, but otherwise the time course, and much of the qualitative content, is quite similar. The effectiveness of this base and of the 4-propoxy-homolog (17; see Section 2.1.8) is 5 to 10 times more than that of mescaline itself and approaches that seen for the 2,4,5-trisubstitution patterns in the substituted phenylisopropylamines, This suggests that the bases that are trioxygenated and presumably indifferent to monoamine oxidase attack may be active independently of whether they are 3,4,5-trisubstituted or 2,4,5-trisubstituted, and of whether they do or do not carry an alpha-methyl group adjacent to the nitrogen atom.
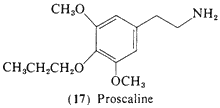
This immediate homolog of (16) is also orally active in man, and some 5-10 times more potent than mescaline, 3,5-Dimethoxy- 4-(n)-propoxyphenethylamine (17, proscaline) shows threshold activity at 15 mg orally and is active in the range of 40-80 mg (Nichols and Shulgin, unpublished data). The trivial names of proscaline (for 17) and escaline (for 16) are suggested by the fact they are related to mescaline by the replacement of a propoxy and an ethoxy, for the methoxy-group of mescaline in the 4-position. The significance of the unusually high activity of this 3,4,5-trisubstituted amine is discussed above.
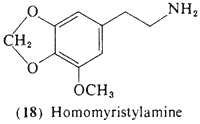
3-Methoxy-4,5-methylenedioxyphenethylamine (18, homomyristylamine, lophophine) is of interest both for biosynthetic and for structural analogy reasons, The compound, although not yet detected per se in the peyote cactus, is a logical intermediate in the biosynthesis of several of the methylenedioxy-substituted tetrahydroisoquinolines known to be present, Although supporting evidence has not been sought. (18) can theoretically participate in the ring-closure reactions with acetaldehyde to form anhalonine (11) and (after N-methylation) lophophorine (10), Furthermore, (18) is the two-carbon analog of the well-established psychotomimetic MMDA (51, Section 3.2.4), The compound has been clinically assayed and is an active psychotomimetic with a threshold level observed at 250 mg (Shulgin, 1976a); thus it has somewhat less than twice the potency of mescaline. Qualitatively it is similar to mescaline in action, with mood elevation progressing into a euphoric state and an enhancement of visual perception, especially in the realm of color. Unlike mescaline, there is little if any nausea and there is no visual distortion.
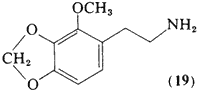
This isomer of homomyristylamine (19, 2-methoxy-3,4- methylenedioxyphenethylamine) has the ethylamine side-chain relocated to a position adjacent to the methoxyl group. It has been titrated to levels in excess of 60 mg orally, acutely. Although there is a pleasant mood elevation reported at this dosage, there are no effects that can be described as psychotomimetic. The reported potency has therefore been recorded as being less than five times that of mescaline (Shulgin et al., 1969).
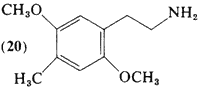
Most of the development of the substitution requirements for activity within the benzenoid psychotomimetics has occurred in studies of compounds with the three-carbon side-chain, The investigation of (20, 2,5-dimethoxy-4-methylphenethylamine) as well as of the halo analogs (21; Section 2.1.12) and (22; Section 2.1.13), was an outgrowth of the recognition of the ring substitution patterns, which afforded highly active psychotomimetic drugs. The corresponding phenylisopropylamines (69, 82, 86) are discussed in Section 3.4, on the three-carbon psychotomimetics.
becomes passive and relaxed and is aware of an integration of sensory perception with emotional state.,. There is a considerable euphoria with an increased body awareness and an increasased receptiveness of visual, auditory, olfactory, and tactile sensation. The integration of sensory and emotional states induces in most subjects a feeling of security and an ability to cope with incidents and experiences that might have led, with drugs such as LSD, to a state of anxiety and possible panic (Shulgin and Carter, 1975).
Six hours following the start of the experiment, the subject is alert, relaxed, and content, with no residual subjective signs of intoxication (Shulgin and Carter, 1975).
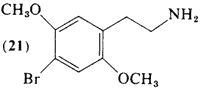
The ethylamine analog of DOB (82) is 2,5-dimethoxy-4-bromophenethylamine (21). It was prepared and assayed in normal subjects (Shulgin and Carter, 1975) following reasoning analogous to that discussed for (20). With a threshold dosage of about 4 mg and an effective dose range of 8-10 mg, it is the most potent of the known ring-substituted phenethylamines. The duration of action (6-8 hr) is somewhat longer than that reported for (20), and at equivalent dosages (based upon threshold values) somewhat less intense. With increasing dosage levels, the intensity but not the duration of the intoxication increases, unlike the homologous phenylisopropylamine (82; Section 3.5.4), where higher dosages led to prolonged residual subjective responses.
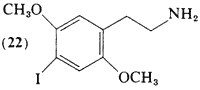
2,5-Dimethoxy-4-iodophenethylamine (22) has been prepared by the iodination of N-(2,5-dimethoxyphenethyl)-phthalimide with ICI followed by the regeneration of the free amine with hydrazine (Braun et at., 1977). Body-distribution kinetics with 131I-labeled material and 123I-labeled material (Braun and Shulgin, 1976) have been compared with the considerably more potent homolog (86) (Sargent et at., 1977), In limited human titrations of (22), threshold central effects are clearly noted at oral levels of 8 mg, but appear to be of short duration. Effective intoxication levels have not yet been explored.
These 13 compounds, all with the intact two-carbon chain and varying only in the position and the identity of ring substituents, are all that have been assayed in human subjects as psychotomimetics. A great number of additional analogs and homologs are known in the scientific literature. As an example, the complete collection of methoxylated phenethylamines (from the mono- to the penta- and with all possible orientation patterns) have been prepared and studied biochemically by Clark et at. (1965), They report that compounds with high degrees of methoxylation serve less well as substrates for enzymatic deamination, but that mescaline (1) was the only isomer (of the 20 studied) that showed an enzymatic deamination that was completely inhibited by semicarbazide. The active 2,3,4- and 2,4,5-isomers (12, 13) and the inactive 3,4-dimethoxyphenethylamine (6) were not similarly affected.
A number of in vivo studies have compared mescaline with these and the other analogs discussed here (with DMPEA, 6, see Bueno, 1975; Carlini et al., 1967; Smythies and Levy, 1960; with 2,3,4-trimethoxyphenethylamine, 12, Winter, 1973). The 3- and 4-monomethoxyphenethylamines have been compared to DMPEA and homopiperonylamine (15) (Epstein et at., 1932) and have been found to be excitants in most species studied, At the other extreme it was only the tetra(2,3,4,5)-methoxy- and pentamethoxyphenethylamine analogs that equaled or exceeded mescaline in behavior-influencing potency studies in rats (Smythies et at., 1967a), The mono-, di-, and trisubstituted isomers (other than mescaline) were inactive. Actual brain-level analyses in rats support this, with both mescaline and pentamethoxyphenethylamine entering the CNS more quickly than 2,4,5-trimethoxyphenethylamine (13) (Cohen et al., 1974). A number of methylated and halogenated analogs of mescaline have been studied in the cat and have been found to be highly active centrally (Benington et al., 1958). No animal model covering the simple substituted phenethylamine psychotomimetics can be considered seriously at the present time until more of the known isomers have been assayed clinically.
N-Methylmescaline (23) has been detected as a minor component in peyote (Sp�th and Bruck, 1937), but human trials of the compound at levels in excess of those conceivably encountered in peyote consumption (i.e., 25 mg) have produced neither central nor peripheral effects (Shulgin, 1967, unpublished data). This suggests that (23) does not contribute to the plant's overall pharmacological toxicity.
N,N-Dimethylmescaline (24, trichocerine) has never been observed in peyote, although the 3-O-demethylated homolog is present and has been studied in biosynthetic schemes (Lundstr�m, 1971b), The compound has been reported as the major component of the mescaline-containing cactus Trichocerus terscheckii (Reti, 1939; Reti and Castrill�n, 1951). The fact that both animals and man can, with impunity, drink the fluids from the crushed pulp of this plant has prompted a study into the psychopharmacological properties of trichocerine.
Ludue�a (1935, 1936) in a single acute experiment consumed 550 mg of the trichocerine hydrochloride and noted no effects of a sensory nature, only a slight gastric heaviness. Vojtechovsky and Krus (1967) have reported that this base has less than one-half the potency of mescaline in humans. At doses of up to 800 mg, with one exception, all responses were weaker than those noted for a 400 mg challenge of mescaline, A 400 mg trial with trichocerine via the perlingual route showed a moderate psychodysleptic effect with a one-hour latency (mescaline required two hours with this mode of absorption). The duration of symptoms was proportionally shorter.
Both the N-monomethyl (N-methylhomopiperonylamine, 25) and the N,N-dimethyl homologs of piperonylamine (15) have been studied clinically as antitussive agents (Brown, 1958). Acute dosages of 30 mg appear to be pharmacologically effective, but there is no mention of central side-effects that might be prodromal to psychotomimetic activity.
In conjunction with the study of the presence of DMPEA (6) in human urine, it was observed that exogenously administered chemical was converted in part to N-acetyl-3,4-dimethoxyphenethylamine (26). This compound is pharmacologically active (Friedhoff and Schweitzer, 1968) and has been studied in man, where it is in part 4-O-demethylated (Schweitzer and Friedhoff, 1968), In studies of this demethylation as a possible biochemical measure to distinguish schizophrenic from normal patients, loading doses were administered (Tozman et at., 1972). Oral administration of up to 500mg of the amine salt produced no reported toxic consequences,
N-Acetylmescaline (27) has been reported as a trace component of peyote (Sp�th and Bruck, 1938). It is also a trace metabolite of mescaline in man, appearing in the urine between the fifth and seventh hour following mescaline administration, and accounting for about 0.1% of the administered drug (Charalampous et al., 1966), This research group has explored the action of N-acetylmescaline (27) in normal humans and found it to be largely without effects in the dose range 300-750 mg, orally. At the highest dose explored, there was a report of a mild degree of drowsiness one hour following administration of the chemical.
Peyote is known to contain a number of additional N-methylated and N-acylated derivatives of substituted phenethylamines (see Kapatia and Fayez, 1973), and a great number in addition are known in the chemical synthetic literature, Several of these basic N-alkyl phenethylamines are known to be pharmacologically active in man and have entered the drug literature as clinical pharmaceuticals, but they have not been studied as, nor are they thought to be, psychotomimetics. Most of these compounds have less than three substituents in the aromatic ring and are generally classified as bronchodilators or stimulants,
Although 3,4,5-trimethoxyphenylacetic acid (28) is not a nitrogen-substituted phenethylamine, it is a major (~30%) metabolite of mescaline in man (Charalampous et al., 1964), and interest in this compound stems from a desire to determine if it is an active biotransformation product, or whether its generation should be classified as a detoxification. Trials with 400 mg orally led to a 75% recovery of unchanged compound in urine, but to no observable effects (Slotta and Muller, 1936). Trials in the dose range 350-750 mg also failed to produce either physiological or psychological changes (Charalampous et al., 1964),
In a study of structural analogs of mescaline, Carlsson et al, (1963) prepared the ethanolamine ether β-(3,4,5-trimethoxyphenoxy)ethylamine (28.1) and the N,N-dimethyl homolog. Again, neither compound is strictly a N-substituted derivative of mescaline, but their close structural similarity, and the fact that they have been assayed in man, makes their inclusion here desirable, The toxicity of (28.1) was determined in the mouse to be 500 mg/kg (LD50, i.p.). In a series of human trials (from 10 to 300 mg dosages), (28.1) was found to be without central effects (mescaline was the control drug, at a 420-mg dose).
The N,N-dimethyl homolog of (28.1) is β-(3,4,5-Trimethoxyphenoxy)-N,N-dimethylethylamine (28.2). It bears the relationship to (28.1) that trichocerine (24) does to mescaline. The LD50 of (28.2) in the mouse was 250 mg/kg i.p. Human trials were conducted over the dose range 10-400 mg, without the appearance of any central effects, Mescaline, at 420-mg total dose, served as the control (Carlsson et al., 1963),
The substitution of a methyl group alpha- to the nitrogen atom in phenethylamine gives rise to the compound amphetamine, a powerful CNS stimulant as well as a peripherally active adrenergic agent, The cardiovascular and stimulatory properties were first reported by Alles (1933). The application of this excitatory property in clinical problems of narcolepsy was initiated by Prinzmetal and Bloomberg (1935). The protracted action and the oral activity of amphetamine is associated with the proximity of the methyl group to the amine function, effectively interfering with enzymatic deamination. The three-carbon chain in general increases toxicity, increases stimulation to the CNS, and decreases the purely "sympathomimetic" nature of the two-carbon chain counterpart (Gunn et al., 1939). There is also the introduction of an asymmetric center, permitting the preparation and study of optical isomers. By far, the largest subfamily of phenethylamine psychotomimetics known are the alpha-methyl phenethylamines, or phenylisopropylamines. As will be indicated they are, as psychotomimetics, in general more potent than their two-carbon counterparts, they are long-acting, and they are orally effective,
One form of psychotogenic action is known that is directly ascribable to the use of amphetamine itself. This is the "amphetamine psychosis" that results form the chronic use of large doses of amphetamine (Monroe and Drell, 1947; Connell, 1958), As the symptoms of stimulation become lost due to the development of tolerance, there is revealed a psychotic state, clinically similar to spontaneous schizophrenia (Bell, 1965). The amount of drug required to evoke this response varies widely from individual to individual (Griffith et al., 1970) but it seems not to depend upon any previous history of predisposition to mental illness (Angrist and Gershon, 1970), This "amphetamine psychosis" has been observed following the chronic abuse of related sympathomimetic stimulants (Greenberg and Lustig, 1966; Angrist et al., 1970a). The psychotropic syndrome that follows chronic drug exposure lies outside this review and will not be included within the concept of psychotomimetic action.
A caution is appropriate concerning the popular custom of referring to this family of α-methyl phenethylamines as "psychotomimetic amphetamines." The name amphetamine designates one unique chemical and there can be no justification for its use in the plural (Shulgin, 1976b). The pharmacologist will consider the stimulant action of amphetamine and will associate it with other sympathomimetics or anorexogenics. A forensic chemist will consider the legal classifications and will probably limit his grouping to the two proscribed drugs amphetamine and methamphetamine, The synthetic chemist will envisage the phenyl ring and the three-carbon chain with the nitrogen on the beta-carbon, whether the compound is biologically active or not, The term will be avoided in this chapter, and the inoffensive substitute "phenylisopropylamines" will be used for this family, Nonetheless, many of the compounds to be discussed in this section have commonly encountered code-names that include a final "A" from this often-used family designation.
These phenylisopropylamines will be grouped in five subsections, according to substitution patterns:
4-Methoxyphenylisopropylamine (29, p-methoxyamphetamine, PMA) is the simplest of the methoxylated phenylisopropylamines to be classified as a psychotomimetic drug, In animal toxicology and pharmacology studies it has been found to be a highly toxic stimulant, showing prolonged cardiovascular effects in the dog at only 0,2 mg/kg (Cheng et al., 1974), In the rat behavioral studies employing the Bovet-Gatti profile procedure (Smythies et al., 1967a) PMA appeared to be a potent hallucinogen, second only to LSD (Smythies et al., 1967c). Para-hydroxylation of amphetamine is a major metabolic pathway in the rat (see Chapter 7 on biotransformation) and it was felt that the psychotogen effects following high doses of amphetamine might be due to some O-transmethylase that could relate amphetamine metabolism to PMA toxicity, Analyses of the urines of experimental subjects in which the amphetamine psychosis had been induced by protracted chronic amphetamine exposure showed no trace of PMA (Angrist et al., 1970b). It has been shown that upon the intentional administration of PMA to human subjects there is a small erratic excretion of the base in unchanged form (Schweitzer and Freidhoff, 1970; Schweitzer et al., 1971). 4-Methoxyphenylisopropylamine has been isolated from monkey brain and CSF following amphetamine administration (Andreoli, et al., 1973). The free hydroxyl counterpart of (29) has been employed therapeutically under the brand name Paredrine as a sympathomimetic in patients with heart block or postural hypotension (Anon., 1973a). Cumulative daily doses of 400 mg are known, and acute dosages of 80 mg produce no central effects related to alertness or mood (Alles, 1959). A recent study in man describes the intravenous administration (acutely) of 2 mg, again without the report of any central effects (Severs et al., 1976). It would appear that PMA is not a contributary agent in the mechanism of the generation of the amphetamine psychosis.
PMA (29) is a treacherous drug to study in human subjects, and its indiscriminate use has been implicated in a number of tragic accidents. The compound has an unusually steep dose-response curve in man. At dosages of 40 mg or less, it is without either peripheral or central effects, Yet at 60-80 mg, the effective dose for the induction of a psychotomimetic syndrome (Shulgin et al., 1969), there have been incidents of precipitous hypertension and cardiovascular stimulation (Angrist, 1969, personal communication), The psychotomimetic state is realized quite suddenly about an hour following ingestion of the drug, and the plateau of central intoxication is passed within the second hour. The somatic effects can persist longer, with blood-pressure elevation, Paresthesia can still be reported four hours following administration.
In early 1973, a flood of PMA appeared in the illicit drug market, first in Canada and then in the United States primarily in the Midwest and the South. A number of deaths have been connected with the use of the drug (Cimbura, 1974) and the procedures for scheduling PMA as a controlled substance were quickly and successfully put into motion (Anon., 1973b), The toxic symptomology associated with overdosage were agitation, respiratory depression, choking, hypertension, extreme hyperthermia, and convulsions, One death occurred 2.5 hr following a parenteral administration, but in the surviving cases the symptomatic crisis was passed at about 8 hr following drug use. No accurate dosages could be determined in these cases, but based upon forensic analysis of evidence associated with hospitalization and autopsy it appears as if as little as 150 mg might be fatal in man. This would represent an extraordinarily small therapeutic index of about 2.5.
The positional isomers of PMA have been studied in experimental animals and appear to be less potent than, and pharmacologically distinct from, PMA (Tseng et al., 1976; Menon et al., 1976). They have never been clinically explored in man, but since they are positional isomers of PMA they are explicitly included in the Federal Drug Schedules. Their physical and chromatographic characteristics have been compared for possible forensic use (Bailey et al., 1974a).
The most interesting of the six possible dimethoxyphenylisopropylamines is the 3,4-isomer (30, DMA, 3,4-DMA, 3,4-dimethoxyamphetamine), since it has the substitution pattern of the neurotransmitters dopamine and norepinephrine, and it is the immediate homolog of DMPEA (6). Unfortunately there have been no clinical studies reported that concern this chemical as a possible psychotomimetic, and very little is known about its intoxicating character, Alles conducted self-experiments in 1962 and "on the basis of threshold effects judged this compound to be two or three times less active than MDA" (Fairchild et at., 1967), Some details are provided in an unpublished work reported by Fairchild (1963): "Oral doses from 10 to 120 mg were without peripheral or subjective effects, except for a slight gastrointestinal discomfort at the higher dose, When 160 mg was ingested a 20 mm of mercury increase in blood pressure occurred within 45 minutes, which was accompanied by a slight mydriasis, lacrimation, and gastro-intestinal uneasiness."
The only account of hallucinogenic effects ascribable to 3,4-DMA (30) is in an Army Chemical Center report (Fairchild, 1963). This group authorized the study of DMA with several psychiatric patients in the New York State Psychiatric Institute:
One patient received 0,004 mM/kg of the hydrochloride salt intravenously (perhaps 70 mg) and exhibited only a slight increase in psychiatric symptoms; a comparable dose in a second individual also elicited only insignificant changes. When one of these two patients was reinjected at a later date with approximately 0.04 mM/kg of 3,4-DMA (perhaps 700 mg i.v.) a definite "mescaline-like" state was induced. The symptoms included colored hallucinations of geometric figures and occasional structured forms, The other individual experienced visual distortions, notable after-imagery, feelings of unreality, and paranoid ideas. Marked mydriasis and gross body tremors also occurred but apparently no hallucinations were experienced.
From these various comments one may assume that the effective dose of 3,4-DMA is approximately that of mescaline, or perhaps 300-400 mg.
There is only a single report of the human effectiveness of 2,4-Dimethoxyphenylisopropylamine (31, 2,4-DMA, 2,4-dimethoxyamphetamine) as a psychotomimetic compound (Shulgin et al., 1969). The potency of five times that of mescaline is based upon acute trials of approximately 60 mg (as the hydrochloride salt, orally), which led to a short-lived intoxication with a considerable component of amphetamine-like stimulation, The euphoria phase peaked at about two hours following administration of the drug, and had largely receded in another hour. Nothing is known of either its metabolism or excretion in the body.
This third positional isomer of dimethoxyphenylisopropylamine (32, 2,5-DMA, 2,5-dimethoxyamphetamine) has a potency in man of some eight times that of mescaline (Shulgin et al., 1969), being effective as an intoxicant at an oral dose of about 50 mg, At this level the drug's effects are closer to a stimulant than a psychotomimetic, There is a sudden onset of action just over an hour following ingestion, which is characterized by vertigo, a closed visual field, and slight muscular incoordination (Shulgin, 1963, unpublished data), Over the following 2 hr there is a modest central intoxication, which is accompanied by a slightly elevated blood pressure and extreme hyperactivity, The syndrome is dissipated at 5 hr.
2,5-DMA appeared in the illicit drug market in 1970 both in Canada (Bailey et al., 1974b) and in the United States (Shaler and Padden, 1972), It was initially misrepresented as mescaline or MDA, but was evemually called by the code name DMA, (In the forensic literature the unprefixed term DMA refers to the 2,5-isomer; in the medical and scientific literature, DMA is assumed to stand for the 3,4-isomer). Seized capsules were found to contain 200 mg of the hydrobromide salt of a high degree of purity (De Zan, 1971) and this must be assumed to have been the dosage employed. This is equivalent to some 170 mg of the hydrochloride, and represents a regimen that has not been studied under controlled clinical conditions.
Although 2,5-DMA has no medical utility and has thus been classified as a Schedule I drug by the Drug Enforcement Administration, there is a considerable demand for it as a chemical in the photographic industry, The manufacturing quota for it, for a single year's production, is 45,000,000 g as the free base (Anon., 1976), and this magnitude of commercial production, in addition to the inexpensive availability of the synthetic precursor 1-(2,5-dimethoxyphenyl)-2-nitropropene, may have accounted for its appearance in high purity and broad availability in the period prior to its legal proscription. The drug has been only rarely seen in the last two years, although it cannot be determined if this is due to effective security control of manufacture, or to an unpopularity of the nature of the intoxication.
The remaining three positional isomers of the dimethoxyphenylisopropylamines are of unknown action in man. They have been studied and compared spectrophotometrically (Bailey, 1972) and chromatographically (Bailey et at., 1974b).
3,4,5-Trimethoxyphenylisopropylamine (33, TMA, trimethoxyamphetamine) is the first psychotomimetic drug that evolved from the systematic application of the principles discovered in studying the relationships between chemical structure and biological activity, Armed with the known structure of mescaline, the proclivity of most phenethylamines to be of only fleeting activity centrally (due to facile deamination), and the effectiveness of a methyl group alpha- to the nitrogen as a stabilizing factor in central activity, Her (1947) synthesized TMA, His favorable impressions on the euphoric properties of the compound encouraged the Canadian group of Peretz and co-workers (1955) to explore its psychopharmacological nature and to evaluate its potential as a psychotomimetic, In their initial studies of the compound, dosage levels of 50-100 mg were administered to normal subjects, In about an hour a transient headache developed, followed by nausea of short duration, without vomiting, This latter symptom was successfully avoided by pretreatment of the subject (30 min) with 50 mg Dramamine, During the second hour the subjects commented upon a sudden onset of giddiness, followed by an increase in movements and communicativeness, and a decrease in inhibitions, There was a slight loss of motor coordination, hyperreflexia, and a modest rise in heart rate without a change in blood pressure, This phase remained for the ensuing three or four hours, and then fell off rather rapidly. At these levels the experience was described as pleasant, and there were no complaints of anxiety or discomfort aside from the slight initial nausea. The interpretation of their studies at somewhat higher levels is complicated by the use of a stroboscope as a visual irritant or a precipitating stimulus for the generation of hallucinations. At 100-125 mg, there was the generation of structured visualization as well as of geometric patterns, often with considerable coloration, when the subject was exposed to the stroboscope at various photic rates. Only occasional image formation (eyes closed) and perceptual distortion (eyes open) were observed without this device.
This geometric isomer of TMA was first synthesized by Bruckner (1933) and its psychotomimetic properties were first observed some 30 years later (Shulgin, 1964a), 2,4,5-Trimethoxyphenylisopropylamine (34, TMA-2, 2,4,5-trimethoxyamphetamine) was the second of the six possible positional isomers found to be psychotomimetic, and was thus called TMA-2. The remaining isomers (see below) have been numbered in order of their progressive position of substitution. As with TMA (33), TMA-2 is not known to occur in nature, The two most commonly encountered essential oils with a trimethoxy substitution pattern are elemicin (35) (and its conjugated rearrangement product isoelemicin, 36) with the 3,4,5-pattern seen in TMA, and the 2,4,5-trimethoxyphenylpropene asarone (37). The elemicin group, along with safrole and myristicin will be discussed in conjunction with nutmeg intoxication (see Section 3.2). Asarone is widely distributed in the plant kingdom, It is a major component of Acorus calamus from a number of different geographical origins (Guenther, 1952), Its common name is sweet calamus or sweet flag, It is used as a medicine by the indians of northern Canada (Hoffer and Osmond, 1967) where it is known by the name rat-root, and is claimed to have intoxicating properties similar to those of LSD, The conversion of asarone to TMA-2 is easily realized chemically but has not been demonstrated as occurring metabolically.
There is a theoretical interest in TMA-2 stemming from the recognition of 6-hydroxydopamine (38) as a potent disrupting agent within the adrenergic nervous system. The two compounds have an identical oxygen substitution, and TMA-2 (34) has been shown to be partially demethylated in vivo (Mitoma, 1970; Sargent et al., 1976), The totally demethylated product from TMA-2 is 2,4,5-trihydroxyphenylisopropylamine (39), which has been explored as an antihypertensive agent, but which exhibits no mental effects at dosages as high as 200 mg (Stone, 1963).
Threshold effects of TMA-2 are evident at dosages of 10 mg as the hydrochloride salt, and the effective oral dose is twice this amount, The drug is therefore some 20 times more potent than mescaline and is the most active of the six trimethoxylated phenylisopropylamines, A generalized presentation of the intoxication state has been presented (Shulgin, 1976c):
The first indications of intoxication usually noted are signs of physical disturbance such as nausea, paraesthesia. and a modest reflexive mydriasis, The central sensory changes appear in the second hour and are characterized by some exaggeration of visual input (especially in the appreciation of colors and contrasts of lighting) and of empathy with irrational objects in one's environment. These preludes lead to a plateau, from three to about six hours following administration, which is an impressive altered state of consciousness virtually free of the distortions and portentousness so common with LSD. The experience dissipates gradually, and is usually completed in 8-10 hours, A sharp dose-response curve exists for TMA-2 in that several additional toxic symptoms have been reported at 25-30 mg levels. There can be a pervasive nausea throughout the entire experimental period, accompanied by actual vomiting, apparent fainting, and brief but repeated periods of amnesia. Peripheral vision can be lost (this, apparently, of hysterical origin) and the accompanying fear of being irreparably severed from reality has led to situations that have proven difficult to manage.
This positional isomer of the trimethoxyphenylisopropylamines (40, TMA-3, 2,3,4-trimethoxyphenylisopropylamine, 2,3,4-trimethoxyamphetamine) is the only one for which no psychotomimetic activity has been observed, A number of acute trials have been carried out at dosage levels of up to 100 mg as the hydrochloride salt (Shulgin, 1964a; Naranjo, 1967, personal communication). At this highest level there were signs of peripheral toxicity, but since there were no indications whatsoever of central disruption further study was discontinued. All of the other trimethoxyphenylisopropylamines had clearly shown some form of sensory or interpretive changes at or below this level; thus it may be stated that TMA-3 is the least active of these isomers, if active at all, In a structure-activity review (Shulgin et al., 1969) the compound was stated to be of activity of less than twice that of mescaline, and this has been occasionally interpreted in animal correlations as indicating the presence of activity but of a low order of potency. Neither statement is exact; the active level of TMA-3, as a psychotomimetic in man, is not known.
TMA-4 (41, 2,3,5-trimethoxyphenylisopropylamine, 2,3,5-trimethoxyamphetamine and the following isomer TMA-5 are extremely scarce compounds, and the extent of the known pharmacology has been obtained on the milligram quantities originally synthesized (Shulgin, 1966a), No peripheral or central effects were noted in several experiments including dosage levels of up to 50 mg, In a single experiment, 80 mg (of the hydrochloride salt) in a drug-sophisticated subject led to an impressive introspective state without any complaint of physical distress, The intensity of intoxication was equated to 50 �g LSD and to 120 mg TMA (Naranjo, 1967, personal communication), If this represents the effective level, then TMA-4 has about four times the potency of mescaline (Shulgin et al., 1969).
As discussed in Section 3.1.8., TMA-5 (42, 2,3,6-trimethoxyphenylisopropylamine, 2,3,6-trimethoxyamphetamine) was available for clinical assay in only small quantities, and there is a corresponding uncertainty concerning the accuracy of the quantitative potency assigned to it. There are initial signs of activity at an oral dosage of 20 mg; these threshold effects include mild mydriasis, increased heart rate, piloerection, and other closely associated prodromal indicators of central activity. A single experimental subject, at 30 mg, underwent a protracted LSD-like session that was adjudged to be similar to the effects produced by 75 mg LSD (Naranjo, 1967, personal communication), With the assumption that this dose represents a valid effective exposure, the drug can tentatively be called a psychotomimetic, and appears to be some ten times more potent than mescaline. The original published value (13x; Shulgin et al., 1969) implies more accuracy than in fact exists, A satisfactory ranking of both TMA-4 and TMA-5 requires much further experimentation.
Human Potencies of the Six
Trimethoxyphenylisopropylamines
Code |
MeO Group Orientation |
Activity (Mescaline = 1) |
TMA-2 | 2,4,5 | 20 |
TMA-5 |
2,3,6 |
10 |
TMA-6 | 2,4,6 | 10 |
TMA-4 | 2,3,5 | 4 |
TMA | 3,4,5 | 2 |
TMA-3 |
2,3,4 |
<2 |
The symmetrical, sixth isomer of trimethoxyphenylisopropylamine is TMA-6 (43, 2,4,6-trimethoxyphenylisopropylamine, 2,4,6-trimethoxyamphetamine), This base was first synthesized over 20 years ago (Benington et al., 1954) and its psychotomimetic properties were discovered 10 years later (Shulgin, 1964, unpublished data), A threshold of central activity is apparent at an oral dose of 20 mg, and the dosage range for a fully developed psychotomimetic intoxication state is 30-40 mg. There is a visually entertaining aspect to the experience, although its extended action (maximum effects can persist into the sixth hour) can be both tiring and anxiety-provoking, Naranjo (1967, personal communication) has investigated TMA-6 with 13 subjects in the 40-80 mg range and has found that these higher doses lead to erratic results, With several patients there was neither a prolongation nor an intensification of effects; and with at least one, there was the generation of a convincing psychotic episode. The recorded potency of TMA-6 (ten times that of mescaline; effective dose about 30 mg) can be used in quantitative comparisons with confidence (Shulgin et al., 1969).
This family of all possible isomers of a single drug, varying widely as it does in comparative human potencies, constitutes an excellent model against which to challenge physical or biological assays for potential psychotomimetic activity. In the several studies that have been reported comparing the human activity of members of this series to physical-chemical properties (Sung and Parker, 1974) and in in vivo animal screens (Uyeno, 1968; Uyeno et al., 1968) there are promising correlations. The best present values for relative human potencies of these six isomers are given in Table 2.
Only one substituted phenylisopropylamine with more than three methoxyl groups has been established as being psychotomimetic, This is 2,3,4,5-tetramethoxyphenylisopropylamine (44, 2,3,4,5-tetramethoxyamphetamine).
A threshold level of central activity is evident at a 30 mg dose orally, and a relatively long-lived disinhibited intoxication is produced by a 50-mg dosage. Compound (44) has been recorded as having six times the potency of mescaline (Shulgin et al., 1969), but additional studies will be needed to establish the qualitative nature of its action.
Although a good correlation has been shown to exist between fluorescence spectra of methoxylated phenylisopropylamines (reflecting molecular orbital energy) and psychotomimetic potency, this isomer (44) and the 2,4,6-trimethoxy analog (43) show a disproportionately feeble fluorescence. Arguments of unusual metabolic protection may be considered in the explanation of the biological activity of these latter two compounds (Antun et al., 1971).
Another potential distinction of these highly methoxylated phenylisopropylamines is the possibility of intramolecular association between an amine hydrogen and an ortho-located methoxyl oxygen that may be undergoing steric displacement from the plane of the benzene ring by interaction with its neighboring methoxyl group (Chothia and Pauling, 1969). The evaluation of the two remaining tetramethoxyl- and the pentamethoxyl-substituted phenylisopropylamines would certainly help resolve these questions.
A second major substitution system found in the alpha-methyl dimethylamine psychotomimetics is the methylenedioxy group. In plants, compounds with this five-membered ether ring are frequently found in close conjunction with the dimethoxy- or the methoxy hydroxy-substituted counterparts. The best known and most thoroughly studied of these essential oils are safrole (45, a structural analog of methyleugenol 46) and myristicin (47, similarly analogous to the already discussed elemicin, 35). Just as there is a close biosynthetic connection between these compounds in plant sources, there is a close interrelationship in the pharmacology of the correspondingly substituted phenylisopropylamines.
There is an extensive literature describing the use of parts or extracts of the plant Myristica fragrans as an intoxicant. Reference should be made to the recent review of Forrest and Heacock (1972) concerning the chemical composition of the plant, to Weil (1967) concerning its psychotomimetic potential, and to Shulgin (1966b) for a discussion concerning the implication of a major essential oil myristicin (47) as a contributing factor to this intoxication. The hypothetical conversion of safrole (45) to MDA (48) and of myristicin (47) to MMDA (51) by the canonical addition of ammonia to the olefinic group (Shulgin et al., 1967) has received some support from recent liver homogenate studies, which indicate chromatographically that substituted phenylisopropylamines can be formed from these olefins (Braun and Kalbhen, 1972, 1973). This could result from the possible formation of a beta-keto intermediate, the corresponding phenylacetone, which is known to be formed metabolically in turn from MDA (Midha, 1974). Oswald et al. (1969, 1971a, 1971b) have found that rats and guinea pigs metabolize both safrole and myristicin via an alpha-keto intermediate, to form amination products through the addition of dimethylamine, pyrrolidine, or piperidine. The pharmacological role that such metabolites might play in an explanation of the intoxicating nature of nutmeg is still only speculative. None of them have been identified as metabolic consequences of human ingestion of nutmeg or mace spices. The phenylisopropylamine analogs have been well explored pharmacologically, however, and will be discussed below under each specific chemical entry.
3,4-Methylenedioxyphenylisopropylamine (48, MDA, 3,4-methylenedioxyamphetamine) is the simplest and the best studied of the methylenedioxyphenylisopropylamines. It was first synthesized by Mannich and Jacobsohn (1910) and first explored in animal studies by Gunn et al. (1939). It was found to be the most effective stimulant of a large group of similarly substituted amphetamine derivatives (including amphetamine).
The initial human trials with MDA were directed toward possible therapeutic relief of Parkinson's disease (Loman et al., 1941) but it was found to be detrimental. Biniecki and Muszynski (1953) observed the analeptic properties of MDA and the levorotatory isomer was clinically explored at Smith Kline and French, Co., as an appetite-suppressing drug (Cook and Fellows, 1961) and as an antidepressant. The compound was found to be equivalent to amphetamine as an anorexogenic agent, but at higher doses (up to 120 mg/day) had central stimulant properties that were found to be objectionable, although hardly classifiable as psychotomimetic (Doughty, 1964, personal communication).
These central effects were explored further by Alles (1959), who following ingestion of 126 mg of the racemate described his subjective changes as follows:
Forty-five minutes (after ingestion) an abundance of curling grey smoke rings was readily observed in the environment whenever a relaxed approach to subjective observation was used. Visually, these had complete reality. When I concentrated my attention on the details of the curling grey forms by trying to note how they would be affected by passing a finger through their apparent field, they melted away. Then, when I relaxed again, the smoke rings were there.
Awareness of the body and of its functioning became subject to a detached special consideration, and the reality of the place of detached observation for a time seemed clearly transposed out of the body and to a place above and to the right rearward. I was compelled to turn my head several times and look - at what part of me could be up there - observing the situation.
At the small, but effective dose level of 150-mg racemic MDA (or 75 mg of the levo- (or R) isomer) the effects are noted within the first hour, and reach their peak quite quickly (in an additional one-half to one hour). The return to the predrug psychological baseline may be quite slow, however, taking as much as an additional 8 hr. The subjective effects generally noted are quite unlike those commonly associated with psychotomimetic drugs. There is little perceptual phenomena, depersonalization, or disturbances of thought, which usually characterize these latter drugs. There is a minimal loss of ability to concentrate on and perform relatively complex visual-motor tasks. Rather, there is an intensification of feelings, a facilitation of self-insight, and the creation of a state of mind that allows increased introspectiveness and insight. There are few objective signs noted of the intoxication state. A small but significant rise in systolic blood pressure occurs in the second to third hour following administration, and a small, insignificant increase in pulse rate during the same period. These latter effects may be due to the dextro- (or S) isomer, since they were noted only in the studies that employed the racemic mixture and not in the levo-isomer studies.
Studies with medium dose levels of the levo-isomer (125-150 mg) appeared to be better suited for psychotherapeutic application (Yensen et at., 1976). There was much more of a tendency to remain within the experience rather than describe it as it was taking place, although communication was fully possible between subject and observer. Externally, the behavior of patients experiencing these doses of MDA more closely resembled that of a similar population under the effects of LSD (200-300 �g). Visions were reported with greater frequency than had been observed with the lower doses. High doses of MDA (again, the levo-isomer, at 200 mg) appeared externally identical to high-dose LSD sessions (300-400 �g). Patients seemed to be absorbed in the unfolding of inner experiences; visions were reported frequently and transcendental-mystical experiences increased in frequency and intensity. There were no reports of distressful physiological problems even with the high dosages employed.
The one possible positional isomer of MDA is 2,3-methylenedioxyphenylisopropylamine. Although it has been listed as an isomer of MDA and as such is a Schedule I drug in the Federal Law Schedule (Anon., 1970), it is at present completely unexplored pharmacologically.
There are only two N-alkyl derivatives of the phenylisopropylamines that have been found to be psychotomimetic in man. Since they are both homologs of MDA, it is appropriate to enter them here, rather than to make a separate section for them. The first is N-methyl-3,4-methylenedioxyphenylisopropylamine (49, MDM, MDMA, 3,4-methylenedioxymethamphetamine), which is structurally analogous to MDA in exactly the same way that methamphetamine is analogous to amphetamine. Two trivial codes have been employed for (49), MDM for the methylenedioxy (MD) and M for the nitrogen substituent, and MDMA, in which the final MA represent methamphetamine. The compound was first reported in the chemical literature by Biniecki and Krajewski (1960) although it was known earlier, having been studied toxicologically by the Army Chemical Center in the 1950s. These studies have recently appeared (Hardman et al., 1973). The compound has had an occasional and erratic appearance in the illicit drug market (Gaston and Rasmussen, 1972; Helisten, 1976, personal communication).
MDM has a higher threshold level than does MDA (48) but otherwise it is very similar in potency. Within the effective dose range (100-150 mg orally) the effects are first noted very quickly, usually within one half-hour following administration. With most subjects the plateau of effects is reported to occur in another one-half to one hour. The intoxication symptoms are largely dissipated in an additional two hours except for a mild residual sympathomimetic stimulation, which can persist for several additional hours. There are few physical indicators of intoxication, and psychological sequelae are virtually nonexistent. Qualitatively the drug appears to evoke an easily controlled altered state of consciousness with emotional and sensual overtones very reminiscent of low levels of MDA (Shulgin and Nichols, 1977).
The second of the N-alkyl phenylisopropylamines known to be psychotomimetic is the ethyl homolog of MDM (49). This is N-ethyl-3,4-methylenedioxyphenylisopropylamine (50, MDE, N-ethyl-3,4-methylenedioxyamphetamine). The compound has made a brief appearance apparently as a synthetic substitute for MDA (Helisten, 1976, personal communication), but it remains substantially undescribed in the scientific literature. MDE has a potency very similar to that of MDM, with 100-125 mg representing an effective dose. The buildup of intoxication is rapid, occurring between one-half and one hour following ingestion, and the effects are already receding before the end of the second hour. Psychopharmacologically this compound is similar to, but slightly faster acting and shorter lived than MDM (Shulgin, 1977, unpublished data). A number of closely related N-methyl homologs of known psychotomimetics have recently been prepared for forensic purposes (Bailey et al., 1975).
The first prepared and first described positional isomer of methoxymethylenedioxyphenylisopropylamine was the 3,4,5-substituted compound MMDA (51, 3-methoxy-4,5-methylenedioxyphenylisopropylamine, 3-methoxy-4,5-methylenedioxyamphetamine).
Three separate lines of structure-activity relationship reasoning led to the preparation and study of MMDA. First, a comparison of 3,4-dimethoxyphenylisopropylamine (DMA, 30) with the methylenedioxy counterpart (MDA, 48) indicated that the replacement of two adjacent methoxyl groups with the five-membered heterocyclic ring diether leads to an unmistakable increase in potency. And similarly, the addition of the third methoxyl function to the amphetamine skeleton (compare DMA, 30, with TMA, 33) also increases potency. Second, a large number of natural alkaloids found in peyote are isoquinolines vicinally substituted with a methoxyl and a methylenedioxy group in the aromatic ring. The possible biosynthetic precursor of these compounds, and the immediate phenethylamine homolog of MMDA (homomyristylamine, 18) has already been discussed. Third, in the natural essential oils, there is a close parallel between elemicin (35) and myristicin (47), both present in the psychopharmacologically active plant nutmeg.
3-Methoxy-4,5-methylenedioxyphenylisopropylamine was synthesized and code-named MMDA, concurrently and independently in two different laboratories (Alles, 1962, personal communication, see Fairchild, 1963; Shulgin, 1964b). The effective dose of MMDA, as the hydrochloride salt, is 120-150 mg and has therefore slightly less than three times the potency of mescaline. A recent monograph has reviewed the known pharmacology and psychopharmacology of MMDA (Shulgin et at., 1973) from which the following generalized description of the objective and subjective symptomology is taken:
In human subjects, the first symptoms appear within 30-60 minutes following administration. Moderate mydriasis was constant, and slight to moderate dizziness was noted by most of the subjects (N = 20). Frequent somatic sensations were those of heat or cold, or trembling. On one occasion (250 mg), a pendular nystagmus was observed in all directions of gaze, and on two occasions a difficulty in focusing was reported. Nausea was present in three subjects for a brief interval, and in one it led to actual vomiting.
The psychological effects were mild, so long as the experience was allowed to develop spontaneously. The phenomena most frequently reported were the accentuation of feelings (anxiety, euphoria, loneliness, loving warmth), the visualization of images (with eyes closed), a state of drowsiness and muscular relaxation, and an overestimation of elapsed time. The imagery was generally realistic, and related to everyday perception of people, landscapes. or objects. The effects usually reached a peak after the first hour following the initial symptoms, diminishing during the second hour, and had disappeared by the end of the fifth hour.
The psychotherapeutic potential of MMDA has been recently explored at great length by Naranjo (1973), who reports numerous patient situations, and compares MMDA with several other psycholytic therapeutic drugs (MDA, harmaline, and ibogaine). There are, as of the present time, no reported studies on the human pharmacokinetics or metabolism of MMDA.
Although not strictly a methylenedioxy derivative, 3-methoxy-4,5-ethylenedioxyphenylisopropylamine (52, MEDA) is the six-membered ring homolog of MMDA and is logically mentioned here. It was reported (Shulgin, 1964b) to be of "marked decrease in psychotropic effectiveness" in comparison with MMDA (51). Acute trials have been extended to oral levels of 200 mg, without indications of central effects. This is three times the level at which initial threshold effects are reported for MMDA, indicating that there is a decrease in psychotomimetic effectiveness with the relief of strain of the heterocyclic ring (from five atoms to six). The seven-membered homolog 3-methoxy-4,5-trimethylenedioxyphenylisopropylamine has been described (Shulgin, 1964b) but it too has not yet been shown to be psychotomimetic.
Most subjects experience threshold effects following oral administration of 15-20 mg of the amine hydrochloride. An effective psychotomimetic intoxication is reported with dosages of 30-50 mg, indicating a drug potency some ten times that of mescaline. The qualitative nature of the psychotomimetic syndrome for most subjects is similar to that reported for MDA. Rather than the anxiety and restlessness common with TMA-2 (the methoxyl counterpart), there is a highly humored empathy and pleasant relaxed state, which persists for 2 to 4 hr. With subjects who appear to have a lower sensitivity to psychotomimetic drugs, the effects have been reported to be paradoxically quite unpleasant. Physical complaints include stomach cramping and intermittent nausea extending well into the period normally free of somatic distress. The psychological effects in this latter subgroup are generally reported as unpleasant and acutely distressing. As with the remaining isomers to be discussed, this drug will have to be much more extensively studied before its psychopharmacological character can be considered established.
There are two positional isomers in the methoxy methylenedioxy series of substituted phenylisopropylamine series that can correspond to the substitution pattern seen in TMA-3 (40). To maintain a consistency of nomenclature between the two parallel families of compounds, the isomers within the 2,3,4-substitution patterns (54, 2-methoxy-3,4-methylenedioxyphenylisopropylamine, MMDA-3a, 2-methoxy-3,4-methylenedioxyamphetamine; and 55 to be discussed below) the first has been suffixed as MMDA-3a and the second as MMDA-3b. There cannot be a 2,4,6-isomer that would correspond to TMA-6 (43).
The initial report that described MMDA-3a clinically and pharmacologically (Shulgin, 1964a) described it as being similar to mescaline in that there was hallucinatory synthesis and total recall, but that it was effective at 16 mg of the hydrochloride salt taken orally. Subsequent clinical study has shown (Shulgin et al., 1969) that in most subjects perhaps twice this quantity is needed to achieve a consistency of imagery (and related phenomena such as slowing of subjective time and a generalized empathy), thus prompting a reassessment of the dosage to be accepted as effective as twice this. MMDA-3a has therefore some ten times the potency of mescaline, but is qualitatively very similar. A note of caution should be added here, in that two subjects (in a study of nine patients) developed a delayed psychotic state, with one unsuccessful suicide attempt.
4-Methoxy-2,3-methylenedioxyphenylisopropylamine (55, MMDA-3b, 4-methoxy-2,3-methylenedioxyamphetamine) is the second of the two possible methoxy methylenedioxy substitution arrangements in the phenylisopropylamine series with the three oxygens substituted adjacent to one another and adjacent to the aliphatic side chain. A single study of its psychotomimetic effectiveness has appeared (Shulgin et al., 1969). Threshold activity grossly similar to that reported for MDA (48) was reported at oral doses of 60 mg of the amine hydrochloride, which would presumably extrapolate to an intoxicative potency of about three times that of mescaline. The qualitative nature of MMDA-3b at such levels must await further clinical studies.
Human Potencies of the Six Methoxy
Methylenedioxyphenylisopropylamines
Code |
Substituent Orientation |
Activity (Mescaline = 1) |
|
MeO |
OCH2O |
||
MMDA-2 | 2 |
4, 5 |
10 |
MMDA-3a | 2 | 3, 4 | 10 |
MMDA-5 | 6 | 2, 3 | 10 |
MMDA | 3 | 4, 5 | 3 |
MMDA-3b | 4 | 2, 3 | 3 |
MMDA-4 | 5 | 2, 3 | ? |
Equally sparse human pharmacology has been reported for 6-methoxy-2,3-methylenedioxyphenylisopropylamine (56, MMDA-5, 6-methoxy-2,3-methylenedioxy- amphetamine) the methylenedioxy analog of TMA-5. Its oral activity is realized at dosages of 30 mg (LaBerge, cited in Shulgin, 1973), which would suggest a potency perhaps ten times that of mescaline.
The sixth possible positional isomer within the MMDA series is the compound 2,3-methylenedioxy- 5-methoxyphenylisopropylamine, MMDA-4, which would correspond in structure to 2,3,5-trimethoxy- phenylisopropylamine, TMA-4 (41). At present, there has been no reported successful synthesis of this compound and, of course, no pharmacological evaluation. The human potencies of these MMDA isomers are ranked in Table 3.
There are six aromatic substitution patterns theoretically possible for the dimethoxymethylenedioxy groups, and three possible orientations for trimethoxymethylenedioxyphenylisopropylamine. A number of these products have been synthesized (see Dallacker, 1969, for leading references). Only two of these compounds have been established as being psychotomimetic in man.
The first, 2,5-dimethoxy-3,4-methylenedioxyphenylisopropylamine (57, DMMDA, 2,5-dimethoxy- 3,4-methylenedioxyamphetamine), bears a relationship to the natural essential oil apiole (2,5-dimethoxy-3,4-methylenedioxy-1-allylbenzene, a major constituent of parsley seed oil, and commonly called parsley camphor) in exactly the same manner that MMDA is related to myristicin. Threshold effects are evident at oral dosages of 20 mg, and twice this amount provokes a psychotomimetic intoxication. There is an abrupt precipitation of effects approximately one hour following drug administration (there is no preliminary notice in the form of nausea, muscular tremors, etc.) and the chronology and subjective nature of the experience closely follow that of LSD. The maximum intensity of the subjective effects occurs from 2 to 4 hr following ingestion, with extensive interpretive distortion, subjective time lengthening, and inappropriate paranoia, followed by a gentle recovery that is largely complete at 8 hr. (Shulgin and Sargent, 1967). The compound thus has some ten times the potency of mescaline.
The second of the isomers that has been studied in man is 2,3-dimethoxy- 4,5-methylenedioxyphenylisopropylamine (58, DMMDA-2, 2,3-dimethoxy-4,5-methylenedioxyamphetamine). Its substitution pattern is that of the essential oil dillapiole, a major component of dill oil. Limited clinical trials have shown that threshold responses are elicited by the oral administration of 30-50 mg of the amine hydrochloride (Shulgin and Sargent, 1967) with the prodromal indicators of an intoxication similar to that of MDA. If the effective dose proves to be twice this, DMMDA-2 may be quantitatively ranked as having about five times the potency of mescaline. Neither of the dimethoxymethyledioxyphenylisopropylamines has been studied metabolically.
Almost all aliphatic ether groups that are found in living processes involve the methyl group. In the biosynthetic pathways that are now quite well understood, there are a number of transmethylase processes known to provide a methyl group to a hydroxy function. In the metabolic chemistry of norepinephrine and epinephrine there is an O-methylation (through the action of the enzyme system catechol-O-methyl transferase, COMT). This is considered to be a possible origin of DMPEA (6) if the latter can be established as being of endogenous origins. Interest in the specific function of the methoxyl groups per se in the substituted phenethylamines stems from two possible interpretations of research in the area.
a. Processes of O-Methylation. One of the principal motives for the research directed to the study of the structure-activity relationships in the area of the psychotomimetics is that there might be an endogenously produced chemical, normally not present or at least not present in sufficient quantity to be effective, which becomes generated or mobilized in examples of spontaneous schizophrenia. It is hoped that some indication of the responsible chemical might become apparent by the comparison of hypothetically generatable natural phenolic biochemicals to known active methoxylated psychotomimetics. Positions that might be the sites of in vivo methylation would be just those positions that would be found to have a structural specificity for a methoxyl group in the exogenous drug.
b. Processes of O-Demethylation. The complement of the above argument, of the generation of a psychotomimetic-like drug by unexpected methylation within the body, is the generation of a product that resembles a normal biochemical by the demethylation of an administered drug. As has been discussed in the section on biotransformation, there is extensive evidence that several of the known psychotomimetics [such as mescaline (1), TMA-2 (34) and DOM (69)] can and do undergo metabolic demethylation in biological systems. Two experimental challenges to this latter process have been explored. One is to replace the methyl aspect of the methoxyl group with another chemical group that may present the body with a dealkylation problem of considerably different ease, and if the demethylation is an enzymatic demethylase process there might be considerable substrate specificity shown. The other is to replace the entire methoxyl group with a different functionality, one that cannot participate in the same metabolic transformations as can the OCH3. In either case it has been hoped that the pharmacological consequence of such a chemical maneuver at a given point of substitution would reflect the importance of that position in any explanation for the mechanism of action of the drug in question.
Both approaches have been followed. Section 2 discussed the replacement of adjacent methoxyl groups with the more labile methylenedioxy counterparts. Section 3 will outline the few homologous ethers that have been studied in man. Section 4 will itemize the alkyl, and Section 5 the halo and sulfur substitution products (substitution in place of a methoxyl group) but products that (mostly) still have methoxyl groups present.
Of the many phenylisopropylamines with variations of the ether function that are known, only one with the 3,4,5-trisubstitution pattern is known to be psychotomimetic in man. This is 4-benzyloxy-3,5-dimethoxyphenylisopropylamine (59). In a clinical study with seven subjects, there was a variable threshold level noted at 30-50 mg, and dosages of 150 mg produced an intense experience (Naranjo, 1967, personal communication). Frequently reported were instances of imagery, mainly of reminiscences of past events, and a continuous intellectual turmoil that seemed to persist for several hours. Direct comparisons with the methoxy counterpart (TMA, 33) indicated that on a weight basis it was distinctly more potent, although the subjects comparing the two drugs found them largely indistingnishable.
All seven of the theoretically possible ethyl homologs of TMA-2 (34) have been chemically prepared and characterized (Shulgin, 1968), but only the three monoethoxy examples have been evaluated as psychotomimetics in man. In this series the Ms and the Es have been generated into code names based upon the first letter of the alkoxy group located in the 2-, 4-, and 5-positions, respectively.
The para-ethoxy homolog of TMA-2 is 4-ethoxy-2,5-dimethoxyphenylisopropylamine (60, MEM, 4-ethoxy-2,5-dimethoxyamphetamine). The threshold and effective levels of MEM are quantitatively similar to those reported for TMA-2, i.e., 12 and 20 mg, respectively (Naranjo, 1967, personal communication). In a study employing nine subjects with dosages ranging from 15 to 40 mg, there were consistent reports of color intensification, wavering and flickering in the visual field, and a relatively long-lasting euphoria. There were indications of extrapyramidal tremors, paresthesia, and a slight mydriasis. The effects are quite slow to be apparent (up to 2 hr) and the overall experience is quite lengthy. Qualitatively, the subjective reports likened MEM as being more like MDA than the homolog TMA-2.
The ortho-ethoxy homolog of TMA-2 (34) is 2-ethoxy-4,5-dimethoxyphenylisopropylamine (61, EMM, 2-ethoxy-4,5-dimethoxyamphetamine). Acute trials have been conducted to levels (30 mg of the hydrochloride salt, orally) more than twice those which precipitate threshold effects with either MEM (60) or TMA-2 (34) (Shulgin, 1968). No effects either central or peripheral were noted, indicating that EMM would be less than half as potent as these latter drugs as a psychotomimetic, if indeed it had that action at all.
5-Ethoxy-2,4-dimethoxyphenylisopropylamine (62, MME, 5-ethoxy-2,4-dimethoxyamphetamine) was found to be without activity in man at acute dosage trials of 30 mg of the hydrochloride salt, orally. This is over twice the threshold dose of MEM (60) and TMA-2 (34), indicating that the compound, if it proves to be psychotomimetic, will have less than half the potency of either of these two latter drugs (Shulgin, 1968). Neither the three diethoxy homologs (EEM, EME, or MEE, see Section 3.3.2) nor the triethoxy homolog (EEE) have been clinically evaluated as of the present time.
An unpublished study of the 4-propoxy homolog of TMA-2 (63, 4-(n)-propoxy-2,5-dimethoxyphenylisopropylamine, MPM, 4-(n)-propoxy-2,5-dimethoxyamphetamine) indicates that it has threshold activity at an oral dose level of about 15 mg, as the hydrochloride. It is then approximately as effective, certainly not much less so, than the two lower homologs MEM (60) and TMA-2 (34). The two higher homologs, 4-(n)-butoxy-2,5-dimethoxyphenylisopropylamine and 4-(n)-amyloxy-2,5-dimethoxyphenylisopropylamine (MBM and MAM, respectively) were without any central effects at similar dosages (12 and 16 mg, respectively, orally, as the hydrochloride salts). Too little is known of this homologous series at the present time to generalize as to structure-activity relationships.
The principal psychotomimetic drugs that are alkyl-substituted phenylisopropylamines contain two methoxyl groups in addition. The several amphetamine homologs with aromatic methylation that have been explored clinically have for the most part been investigated as stimulants, as analgesics, or as appetite-suppressing agents. However, in the last few years there have been a number of reports of 4-methylphenylisopropylamine (4-methylamphetamine, 64) appearing as an abuse drug in the illicit market in North Carolina (Keaton, 1973), in Pennsylvania (Cordova, 1974) and in Canada (Bailey et al., 1974a). As several isomers and homologs of these simple aliphatic substituted amphetamine derivatives have been clinically studied in man, they are included in this section along with the analogs containing methoxyl groups.
The sporadic appearance of 4-methylphenylisopropylamine (64, p-tolylisopropylamine, 4-methylamphetamine, Aptrol) in the illicit market (see references above) may be due to its possible pharmacological effectiveness as a stimulant and as a physically and emotionally disruptive drug. It has been clinically studied as an anorexogenic by Smith Klein and French Laboratories under the name Aptrol. At 75 mg doses, in normal subjects, this effect is generally recognized, without any expression of central complications. Marsh and Herring (1950) have observed nausea, sweating, and blood pressure elevation at higher levels. At about 150-mg dosages orally (as the sulfate) there were overt signs of toxicity (gastric distress, salivation, coughing, vomiting), which effectively masked any expression of central stimulation common with amphetamine. This behavioral display is completely compatible with that found in rats with the Bovet-Gatti analysis (Beaton et al., 1968) wherein 4-methylphenylisopropylamine, unlike 4-methoxyphenylisopropylamine, appeared to be a weak stimulant rather than a hallucinogenic. The decrease in pressor effectiveness found in the phenylisopropylamine series by the addition of the 4-methyl group (64 vs.amphetamine) parallels dog studies with the corresponding phenethylamines (Hambourger and Jamieson, 1936).
As of the present time, there are no reports of the dosages available in illicit use, nor of the pharmacological effects experienced.
Only limited studies have been made in the human evaluation of 2-methylphenylisopropylamine (65, o-tolylisopropylamine, 2-methylamphetamine) and these have been directed toward its evaluation as a potential anorexogenic agent (Marsh and Herring, 1950). Although there were some indications of pressor effectiveness at as little as 40 mg orally (as the sulfate), at 150-mg dosages these changes were no more than those induced by less than half this dosage of control amphetamine, and were less than those induced by similar doses of either the 3- or 4-positional isomers (compounds 66 and 64). The only evidence of mood change was an increase in talkativeness at this highest level.
The meta-isomer of the tolylisopropylamines (66, 3-methylphenylisopropylamine, m-tolylisopropylamine, 3-methylamphetamine) has been clinically explored as a possible anorexogenic agent by Marsh and Herring (1950). Orally administered doses of (66) (as the sulfate) of up to 150 mg produced a cardiovascular response similar to the 4-methyl isomer (64) at similar dosages, but the subjects seem to be relatively free of the distressful side-effects produced by this latter drug. Some slight evidence of central nervous system stimulation was reported.
Interest has been directed toward 3,4-dimethylphenylisopropylamine (67, xylopropamine, Perhedrin, Esanin) as either an analgesic or an anorexogenic agent. At acute dosages of 10 mg orally (as the sulfate) there was some relief in experimental subjects to electrically induced pain (via electrodes to tooth fillings), without any central changes noted that could be considered in any way as being stimulant or psychotomimetic (Harris and Worley, 1957). No cardiovascular effects are noted in man until levels of about 100 mg are administered (Marsh and Herring, 1950), and at 150-mg dose levels there is a relatively short-lived toxicology picture of nausea, vomiting, and reported collapse.
A single report has been published concerning the effects of 2,5-dimethylphenylisopropylamine (68) in man (Marsh and Herring, 1950). In studies directed toward the possible anorexogenic effectiveness of homologs of amphetamine, they found that even at levels of 150 mg (of the sulfate, orally), (68) had very little obvious activity on either the cardiovascular system or the central nervous system of man.
A number of positional isomers of the Ar-polymethylated phenylisopropylamines have been chemically described [the 2,4-di- (Anon., 1959); 2,4.6-tri- (Buu-Hoi and Petit, 1960); 3,4,5-tri- (Benington et al., 1958)]. These chemicals have been explored pharmacologically in experimental animals but there are no reports of their action in man.
In contrast to the relative cardiovascular and central inactivity of the simpler methylated homologs of amphetamine, it is now known that the addition of methoxyl groups to the aromatic ring can result in compounds that are not only highly potent, but which are psychotomimetic as well. The first member of this family, and one of the most completely studied, is 2,5-dimethoxy-4-methylphenylisopropylamine (69, DOM, STP).
The rationale for its synthesis and pharmacological study was based upon the suspected participation of the aromatic 4-position in the mechanism of action of several of the phenethylamine psychotomimetics. The replacement of the potentially labile methoxyl ether with a hydrolytically stable but oxidatively vulnerable methyl group (compare TMA-2, 34, with DOM, 69) led to a compound with a severalfold increase in human potency, and one with a considerably extended course of action. Threshold effects can he recognized at oral levels between 1 and 2 mg. There is a generalized awareness of minor physical disturbances (muscular tremor, facial flushing, paresthesia) that occur about an hour following these low levels of the drug's administration, and marginal sensory amplification (pleasure from sights and smells, a relaxed contentment), which give indications of the long chronology to be expected with increased dosage.
DOM was first synthesized and its psychotomimetic properties discovered in 1963 (Shulgin, 1963, unpublished data). Its initial appearance within the drug abuse population occurred in San Francisco in mid-1967, with the distribution of thousands of tablets, leading to a pandemonium of acute toxic reactions (Smith, 1969). The weight of DOM in the dosage unit distributed was 10 mg, although initially a small distribution was made of units of twice this quantity (Meyers et al., 1968). Two controlled clinical studies with normal subjects were quickly conducted (Snyder et al., 1967), which were directed both toward a characterization of the drug at low doses (Snyder et al., 1968; Faillace et al., 1970; Weingartner et al., 1971) as well as toward a study of the nature of the intoxication induced by higher doses (Hollister et al., 1969). There appears to be a dose-dependent biphasic response to the drug. At dosage levels between 2 and 5 mg, the Snyder group reported their subjects to be substantially free of objectively observed physiological change, and of perceptual distortion. There were clear indications of abnormal responses that depended upon intact cognition and the interpretation of visual signals, but there were no indications of effects that they could call either hallucinogenic or psychotomimetic. The peak of central activity in these studies was between 3 and 4 hr on the average.
In studies involving higher dosages (to 14 mg, acutely, orally) the Hollister group observed extensive somatic as well as perceptual and psychic changes (see Hollister it al., 1969, for details and chronology). At the time of the extensive street appearance of DOM, it was thought that chlorpromazine, rather than ameliorating the drug's action, actually aggravated the psychotic aspects of the intoxication. This point was specifically challenged by the administration of therapeutic amounts (50-200 mg) of chlorpromazine to experimental subjects concurrently with large doses of DOM. There were no instances of complete reversal of the toxic syndrome, but the sedating action appeared generally to attenuate some aspects of the intoxication.
Two studies have been made of the development of tolerance to DOM by repeated exposure. Hollister et at. (1969) administered gradually increasing doses to his subjects until a total of 12 mg had been administered on a single day (4 mg, t.i.d.); on the following day the subject was challenged with a single 12-mg dose. Angrist et al. (1974) administered 6 mg per day for three consecutive days. Both groups reported the development of a high degree of tolerance, as judged both by objective measurement and by subjective evaluation.
The pharmacology of the individual R and S isomers of DOM has been studied. Animal behavioral tests (Benington et al., 1973) as well as in vitro studies of serotonin agonism activity (Dyer et at., 1973) have indicated that the R or levorotatory isomer should be the major contributor to the psychotomimetic activity of the racemate. Human studies (Shulgin, 1973b) have reported this same conclusion.
The biotransformation fate of DOM has been investigated in several systems. The major metabolite observed is the benzyl alcohol resulting from the oxidation of the 4-methyl group. In in vitro preparations, this hydroxylation product is the major derivative observed, although there is evidence of a minor metabolic process involving O-demethylation (Weinkam et al., 1976). If there were bis-O-demethylation of DOM, the product would be a hydroquinone that is known to spontaneously air-oxidize to a potentially bioactive indole (Zweig and Castagnoli, 1974). The hydroquinone was found (Zweig and Castagnoli, 1975) but appeared not to be further oxidized in the in vitro system employed. Body distribution of DOM and its metabolites has been studied in a number of animal species (Id�np��n-Heikkil� et al., 1969, 1970; Id�np��n-Heikkil� and McIsaac, 1970; Ho et al., 1971a).
The only positional isomer of DOM (69) that has been explored as a psychotomimetic in man is 2,6-dimethoxy-4-methylphenylisopropylamine (70, Z-7). Its threshold of central activity is realized at 10-15 mg orally, as the hydrochloride, indicating that (70) may be slightly more potent than the methoxy counterpart TMA-6 (43). The duration of action is also slightly longer (up to 8 hr). Insufficient clinical studies have been completed to assign a probable full activity dosage requirement as yet.
Isomers of Methoxy Methyl Phenylisopropylamine
| 2 | 3 | 4 | 5 | 6 | Reference |
| Disubstituted | |||||
OCH3 | CH3 | Horii and Inoi (1957) | |||
OCH3 | CH3 | Ho et al. (1970b) | |||
OCH3 | CH3 | Horii and Inoi (1957) | |||
OCH3 | CH3 | Morishita et al. (1956) | |||
CH3 | OCH3 | Carlsson et al. (1970) | |||
OCH3 | CH3 | Ho et al. (1970b) | |||
CH3 | OCH3 | Wenner (1951) | |||
CH3 | OCH3 | Carlsson et al. (1970) | |||
| Trisubstituted | |||||
CH3 | OCH3 | CH3 | Burger and Foggio (1956) | ||
CH3 | OCH3 | OCH3 | Anderson et al. (1977) | ||
OCH3 | CH3 | OCH3 | Shulgin (1970b) | ||
OCH3 | OCH3 | CH3 | Anderson et al. (1977) | ||
OCH3 | CH3 | OCH3 | Shulgin (19705) | ||
CH3 | OCH3 | CH3 | Ho et al. (1970b) | ||
CH3 | O-CH2-O | Sugasawa and Hino (1954) | |||
A theoretical complication is introduced into the understanding of the mechanisms of action of the psychotomimetic drugs by the activity of compounds such as (43) and (70). One of the arguments advanced to explain the increase in biological activity associated with the 2,4,5-ring substitution pattern has been the theoretical ease with which such compounds can undergo oxidation to para-quinones and possible subsequent indole formation [the prerequisite intermediate hydroquinone in the example of DOM (69) has actually been isolated from in vitro preparations (Zweig and Castagnoli, 1975)]. However, compounds with the 2,4,6-orientation are incapable of such oxidation without the introduction of an oxygen into the aromatic nucleus. A major step toward uncovering a common mode of action of the 2,4,5- and the 2,4,6-trisubstituted psychotomimetics will be taken when and if it can be shown that the metabolic disposition of the latter system involves an additional oxidation step and that the resulting phenols (or their methyl ethers) are biologically active.
A large number of substitutional and positional isomers of the methoxy methyl phenylisopropylamines have been synthesized and chemically characterized, These can be thought of as manipulations and rearrangements of the DOM structure, and should represent the raw material from which an explanation of the mechanism of action will come. Several examples of promising animal pharmacology have been observed with some of these analogs. Ho et al. (1970b) have reported that 3-methoxy-4-methylphenylisopropylamine (71; DOM missing the ortho-methoxyl group and thus unable to form a benzoquinone) was equivalent to DOM itself in both the nature and the duration of this action. This compound has apparently been seen in a street sample seized in Italy, where it had been considered hallucinogenic, with the code initials MMA (De Zorsi and Cavalli, 1974). Anderson et al. (1977) have found that both 2,4-dimethoxy-5-methylphenylisopropylamine (72) and 4,5-dimethoxy-2-methylphenylisopropylamine (73) (DOM with its substituents interchanged to preclude the formation of a benzoquinone by simple demethylation) are inactive in the rabbit hyperthermia assay of Aldous et al. (1974). Table 4 itemizes these analogs with their leading references.
As with DOM (69), threshold effects of DOET are clearly apparent in the 1-2 mg range, when administered orally to normal experimental subjects. In a series of studies employing a 1.5-mg dose (with an active placebo control of 10 mg amphetamine), Snyder and co-workers have reported a generalized chronology of onset at 1-1.5 hr, a peak of effect at 3-4 hr, with subjective symptoms largely subsided after 6 hr (Snyder et al., 1968, 1969; Weingartner et al., 1970). Subjectively the effects are consistently different from the psychotomimetic syndrome observed with higher levels of DOM. There was the generation of a mild euphoria, feeling of enhanced self-awareness, eyes-closed imagery, and changes in the cognitive processes in areas such as free association. In a study involving a range of dosages (from 0.75 to 4 mg) these symptoms appeared consistently, but at no dose level were there indications of hallucinations or of disruptive psychotomimetic toxicity (Snyder et al., 1971). It would appear that the induced behavior changes, at least over this range, are dose independent. In a study of the two optical isomers of DOET, independently and in comparison with the racemate, the R or levo-isomer is perhaps twice as effective as the DL-racemate, which in turn is twice as effective as the S or dextro-isomer (Snyder et al., 1974). There are many problems involved in any attempt to interrelate potencies of two compounds such as DOM (with a narrow range of benign effectiveness, 2-5 mg, and above this level the production of a dysphoric and LSD-like state rather abruptly) and DOET (which shows a largely dose-independent subjective intoxication syndrome over the same range, i.e., up to 4 mg). No hallucinogenic effects have been reported with the oral use of DOET, but then there are no reports of oral use exceeding 4.0 mg. Comparing the effective threshold levels of the two drugs (at which level the induced effects are largely indistinguishable both in nature and in duration) it would appear that DOET is slightly more potent and should be considered as having some one hundred times the potency of mescaline. This would arbitrarily place its effective dose (of the racemate) at about 4.0 mg.
The metabolism of DOET has been studied. In the rat, the principal site of oxidation appears to be the ethyl group in the 4-position (Ho, 1975) with approximately half of the administered dose being excreted as the beta-hydroxymethyl analog, and some 18% as the 4-carboxymethyl oxidation product (Tansey at al., 1975). These were excreted, in part, in conjugated form. Some 14% of the administered amine was excreted unchanged. In human studies there was between 10 and 40% of the free base excreted in unchanged form, with the greatest concentration occurring in the 3-6 hr collection period (Snyder it al., 1969). Limited studies on the kinetics of body distribution of DOET (Ho, 1975) indicate that it is taken up (in the rat) into tissue more rapidly and to a higher level than is DOM, but that DOM is retained longer and is more slowly metabolized. The biochemical and behavioral animal studies of DOET and the related homologs discussed here are entered below DOAM (77).
Fig. 1.
Comparison of the in vitro and the in vivo assay
of a homologous series of psychotomimetics
(4-alkyl-2,5-dimethoxyphenylisopropylamine).
The immediate homolog of DOET, by the elongation of the alkyl chain at the aromatic 4-position, is 2,5-dimethoxy-4-phenyl- isopropylamine (75, DOPR). In analogy with DOET, this compound was initially encoded DOP, and the last letter added at a later time. A potential ambiguity exists in the literature with the use of this code. The term DOP was employed in a study of a compound thought to be the 4-isopropyl isomer (Kulkarni, 1973). It is now known that the compound employed in this study was in fact the n-propyl isomer, and that therefore both DOP and DOPR in the literature refer to compound (75). Recently the isopropyl isomer has been prepared (Aldous et al., 1974) and in animal studies appears to have reduced potency. Nothing is known of its human psychopharmacology.
DOPR (75), as with the two higher homologs (76) and (77), has not been pharmacologically characterized to the same extent as have the methyl and ethyl counterparts (69) and (74). The rat behavioral studies of Morin et at. (1975) as well as the biochemical studies of Shulgin and Dyer (1975) both indicate that the propyl substitution should produce the greatest potency of the entire series of 4-alkyl substituted 2,5-dimethoxyphenyl- isopropylamines (see Fig. 1). In humans, threshold effects are regularly noted in orally administered doses of less than 1 mg, but the effects observed with doses in the 1-2 mg area are generally less extreme than those noted with equal dose level administrations of DOET in control experiments. This suggests that the drug is less effective and should be quantitatively equated with DOM (Shulgin and Dyer, 1975) (see Fig. 1). There is not sufficient information available to determine if the effects of DOPR should qualitatively be classified with DOM or with DOET.
The four-carbon homolog in this series, 2,5-dimethoxy-4-butylphenylisopropylamine (76, DOBU), appears in the animal behavior tests (see DOAM, 77) to be a highly potent compound, although somewhat less active than the three-carbon counterpart. The compound shows clear threshold effects in man in the 1-2 mg area, acutely and orally, and is effective at dosage levels slightly more than twice those required for DOM (69). It has been assigned (Shulgin and Dyer, 1975) a relative potency 36 times that of mescaline, although the qualitative nature has not yet been adequately investigated. As with the 4-propyl counterpart (75) there seems to be a sympathomimetic stimulatory component associated with the effective dosage.
The last member of this series of homologs is 2,5-dimethoxy-4-amylphenylisopropylamine (77, DOAM). As will be seen below, it has been studied both in behavioral tests and in biochemical systems, and in both it appears to be of substantially reduced potency compared with its two immediately lower homologs. Limited human evaluation has provided the same results. Threshold effects are noticed at levels of 5-10 mg orally, and the compound has a stated effective potency of only 10 times that of mescaline (Shulgin and Dyer, 1975). No qualitative description of its intoxication syndrome at effective levels can be made from the limited experimental data available.
As mentioned above, throughout this series both in vivo and in vitro assays have been surprisingly consistent and in good agreement with the reported human values of activity. In the modified Discrimination Sidman Avoidance Schedule animal behavioral test (which was mentioned earlier) Morin et al. (1975) have found the propyl homolog (75) to be the most effective compound in disrupting behavior. followed closely by the n-butyl homolog (76). The 4-methyl and 4-ethyl compounds (DOM, 69 and DOET, 74) were intermediate in effectiveness, and the 4-H and the 4-n-amyl compounds (2,5-DMA, 32, and DOAM, 77) were ineffective. In an in vitro assay for serotonin agonism, these same compounds were ranked in almost precisely the same way (Shulgin and Dyer, 1975). The n-propyl homolog was the most effective agonist, with the other homologs dropping away on either side. The human effectiveness of this family is seen to be greatest at the ethyl compound, with the methyl (DOM, 69) and propyl (DOPR, 75) being equipotent within experimental variability; the remaining isomers are significantly less potent as psychotomimetics. These data are shown in Fig. 1.
In a synthetic study of 4-alkyl-substituted 2,5-dimethoxyphenylisopropylamines a number of compounds were prepared in which the 4-alkyl and the 5-alkoxy groups were tied together in a furan or a pyran ring, thus showing a passing resemblance to the heterocyclic structure of tetrahydrocannabinol (Shulgin, 1971). The title compound (78) has been assayed as a psychotomimetic and is at least one order of magnitude less potent than the isosteric analogs DOET (74) and DOPR (75). There are no detectable effects noted following oral administration in excess of 10 mg, acutely.
The last subdivision of the variously substituted phenylisopropylamine psychotomimetics contains those compounds, largely with methoxyl groups which are substituted in addition with atoms other than carbon or oxygen. The introduction of the halogen into the phenylisopropylamine nucleus has generally been inspired by an effort to interfere with metabolic attack. In the case of the simplest unsubstituted compounds such as amphetamine and methamphetamine, there was a desire to interfere with the known oxidative attack at the aromatic para position. It was hoped to create a molecule that might have some of the pharmacological virtues of amphetamine (anoxia, alerting) without the dependence problems. In the case of the more highly substituted phenylisopropylamines, the effectiveness found to follow the replacement of a para-methoxy group with a para-methyl group [increase in potency comparing TMA-2 (34) with DOM (69)] might be yet further enhanced by replacing this latter group with a metabolically refractory halide atom. The substitution of sulfur atoms in the aromatic nucleus was made to study the effects of isosteres (i.e., the replacement of a methoxy with a thiomethoxy group). Both lines of study have led to compounds of pharmacological interest.
The simplest of the halogenated phenylisopropylamines is 4-chlorophenylisopropylamine (79, para-chloroamphetamine, 4-CA). It and the N-methyl homolog (80) are highly active compounds in experimental animals, producing a remarkably long-lasting depletion of brain serotonin levels (Pletscher et al., 1963) and a decrease in tryptophane hydroxylase activity (Sanders-Bush et al., 1972).
Considerable clinical application of 4-CA has been made, and it has been found effective as an antidepressant when used chronically at levels of 75 mg/day (van Praag et al., 1971; van Praag and Korf, 1976). There are very few side effects noted and the drug is tolerated very well. However, indications of raphe-nucleus degeneration (Yunger et al., 1974) and related neurotoxicity (Harvey and McMaster, 1976) in experimental animals have discouraged further clinical study.
An unusual aspect of 4-CA metabolism is the reported conversion of the drug to oxygen-containing products. A phenolic product was identified by Parli and Schmidt (1975) as being 3-chloro-4-hydroxyphenylisopropylamine. This would seem to invoke the NIH shift as an explanation for the migration of the chloro atom. Even more remarkable is the report (Sherman and Gal, 1976) of the isolation of 3,4-dimethoxyphenylisopropylamine following the intraventricular injection of 4-CA. This represents the formation in vivo of a weak but accepted pressor and psychotomimetic. When the mechanism of its formation is understood, a chemical link may be at hand tying the simpler phenylisopropylamine stimulants to the methoxylated psychotomimetics. There were no reports from the clinical studies of 4-CA that suggested any psychotomimetic action.
The N-methyl homolog of 4-chlorophenylisopropylamine (80, para-chloromethamphetamine p-CMA, Ro 4-6861, S-33) was also found to be a potent and long-lasting depleter of brain serotonin (Fuller et al., 1965). It has been compared with methamphetamine in normal subjects (Verster and van Praag, 1970) and has been evaluated clinically in comparison with 4-CA (79) as an antidepressant (Deniker et al., 1971; van Praag et al., 1971; van Praag and Korf, 1976). Typical dosages were between 60 and 90 mg/day, administered chronically for several weeks. There appeared to be no physical or psychic dependence developed, no cardiovascular complications, and no sleep or appetite problems. There was no mention made of mental disturbances that might be considered psychotomimetic.
The alpha,alpha-dimethylphenylethylamine homologs of p-CMA have been explored clinicaly as anorexics. 4-Chloro-alpha-alpha-dimethylphenethylamine is used therapeutically under the name of Chlorphentermine; the ortho-isomer is known as Clortermine.
The bromo-counterparts of the chlorophenylisopropylamine have been studied, but have not found extensive clinical evaluation. The primary amine 4-bromophenylisopropylamine (4-bromoamphetamine) is, like the 4-chloro-analog 4-CA (79), a long-term depleter of serotonin in the brain (Fuller et al. (1975). The 4-fluoro analog, while still effective biochemically, is not of as long a duration of action. The N-methyl homolog of 4-bromo-phenylisopropylamine has demanded interest from a separate point of view, however. This compound, 4-bromo-N-methylphenylisopropylamine (81, V-111, p-bromomethamphetamine), has been found to give pharmacological profiles in a large number of animal species, which are indistinguishable from those shown by LSD and other psychotomimetics (Knoll et al., 1970). Although much of the literature appearing over the period from 1965 to 1975 refers to (81) as a psychotomimetic, it had apparently never been clinically assayed in man. It is now known that the compound "has no psychotomimetic effect whatsoever in humans" (Knoll, 1974, personal communication). The high pharmacological potency of (V-111) in the biochemistry of serotonin and its apparent enhancement of learning and memory in experimental animals have maintained an active interest in it in the research area.
The addition of methoxyl groups to the nucleus of 4-bromoamphetamine leads to compounds that have proven to be not only psychotomimetic in man, but to be among the most potent phenylethylamine derivatives yet reported in the scientific literature. The first of this family of compounds to be reported was 4-bromo-2,5-dimethoxyphenylisopropylamine (82, DOB. 4-bromo-2,5-dimethoxyamphetamine), which has the substitution pattern of so many of the psychotomimetics discussed earlier, the 2,5-dimethoxy orientation with an additional substituent at the 4-position. DOB (82) shows an active threshold effect at oral dosages of about 0.4 mg and exhibits its full psychotomimetic activity over the range 0.8-2.0 mg (Shulgin et al., 1971). At these higher levels (up to 2 mg) there appeared
to be an increase of both intellectual and emotional stimulation, whereas perceptual enhancement seemed not to be dose-dependent. There was an increase in the level of fluency and attention, while maintaining full communication capabilities. This compound is thus similar to 3,4-methylenedioxyamphetamine (MDA) except that here the subjects were more active and had greater contact with the environment. The sense of added significance in ordinary events and motivation towards introspection were similar to that of the hallucinogens such as mescaline, but with a complete lack of imagery or perceptual distortion.
A second brominated aromatic ether studied by the Cassels groups (Sepulveda et al., 1972) is 2-bromo-4,5-methylenedioxyphenylisopropylamine (84). This is the bromination product of MDA (48) and can be considered the bromo analog of MMDA-2 (53). At dosages of 350 mg orally in man (weight-equivalent to those needed of mescaline, but slightly more potent with consideration of molarity) there was the generation of amphetamine-like responses, indicating central activity but with no detail reported to indicate that this activity was psychotomimetic in nature. This can only be resolved with additional clinical study.
Brominated Alkoxylated Phenylisopropylamines
R2 |
R3 |
R4 |
R5 |
R6 |
Reference |
| Disubstituted | |||||
Br | OCH3 | Barfknecht and Nichols (1971) | |||
Br | OCH3 | Barfknecht and Nichols (1971) | |||
OCH3 | Br | Barfknecht and Nichols (1971) | |||
| Trisubstituted | |||||
Br | OCH3 | OCH3 | Bailey et al. (1976) | ||
Br | OCH3 | OCH3 | Barfknecht and Nichols (1971) | ||
Br | O-CH2-O | Sepulveda et al. (1972) | |||
OCH3 | Br | OCH3 | Shulgin et al. (1971) | ||
OCH3 | Br | OCH3 | Barfknecht and Nichols (1971) | ||
OCH3 | OCH3 | Br | Sepulveda et al. (1972) | ||
OCH3 | Br | OCH3 | Bailey et at. (1976) | ||
Br | OCH3 | OCH3 | Cassels (1976, pers. comm.) | ||
Br | O-CH2-O | Cassels (1976, pers. comm.) | |||
Br | OCH3 | Br | Cassels (1976, pers. comm.) | ||
OCH3 | OCH3 | Br | Bailey et al. (1976) | ||
| Tetrasubstituted | |||||
Br | OCH3 | OCH3 | Br | Bailey et al. (1976) | |
4-Bromo-3,5-dimethoxyphenyl- isopropylamine (85) has been prepared (Barfknecht and Nichols, 1971) and found to be centrally active in man (Nichols et al., 1977). Threshold effects are first noted in the dose range 3�6 mg, and at 10 mg orally there are indications of both mental (psychotomimetic?) and physical (central analgesia) effects. The action of the drug appears to be complex, and a single assignment of its character will have to await additional studies.
A number of additional brominated methoxylated phenylisopropylamines are known chemically, but have not as yet been studied in man.
2-Bromo-5-methoxyphenyl- isopropylamine and
2-bromo-4,5-dimethoxyphenyl- isopropylamine have been found to be inactive in the rat response screening (Barfknecht and Nichols, 1971), which showed DOB (82) to be highly potent. The
study of the fluorescence of several isomers (Antun et al., 1971) has been correlated to animal activity. In a chemical study of the bromination products obtained directly from the
several known dimethoxyphenylisopropylamines, Bailey et al. (1976) have prepared a number of heretofore unknown isomers. The listing of the currently known brominated methoxylated
phenylisopropylamines is presented in Table 5 with appropriate references.
Unlike the bromination products of the methoxylated phenylisopropylamines, the analogs that contain iodine in the aromatic nucleus must be made indirectly. The only iodinated analog known to be psychotomimetic (and also the only iodinated organic molecule known to be psychotomimetic) is 4-iodo-2,5-dimethoxyphenylisopropylamine (86, DOI, 4-iodo-2,5- dimethoxyamphetamine). It has been prepared from the 4-amino analog by the Sandmeyer reaction (with the aliphatic amine function blocked by the acetyl group) (Coutts and Malicky, 1973) or from the 4-hydrogen analog by the action of iodine monochloride (with the amine function blocked with the phthalic acid imide group) (Braun et al., 1977). The compound has been prepared with radioisotopic iodine (both 131I and 123I; Braun et al., 1977) and its distribution studied in experimental animals (Sargent et al., 1977). Human clinical trials have shown the compound to have both the potency and the duration of action of the brominated counterpart (DOB, 82).
The corresponding chloro analog (4-chloro-2,5-dimethoxyphenylisopropylamine) has been synthesized (Coutts and Malicky, 1973) and found to be equivalent to both the bromo (82) and the methyl (69) analogs, but it has not been reported to be psychotomimetic in man. The 4-fluoro analog has not been synthesized.
Sulfur analogs of several of the indolic psychotomimetics have been synthesized and pharmacologically investigated (of DMT, Harrison et al., 1974; of 5-methoxy-DMT, Bosin et al., 1976; of portions of the LSD molecule, Campaigne and Knapp, 1970). A recent report has described the preparation of the 4-thio analog of TMA-2 (Nichols and Shulgin, 1976). This base, 4-thiomethoxy-2,5-dimethoxyphenylisopropylamine (87, para-DOT) is the only one of the three possible positional isomers found to be psychotomimetic. In man, the potency of 87 lies intermediate to the two analogs, which have a methoxy group (TMA-2, 34) or a methyl group (DOM, 69) at the 4-position instead of the methyl-thin group (Shulgin and Nichols, 1977). Its threshold and active levels in normal experimental subjects lie between 5 and 15 mg, administered orally. The chronology of the induced intoxication resembles TMA-2 (34). Initial effects are noted at just over one hour following administration with the maximum effect reached at about the end of the second hour. The plateau is maintained for about 1.5-2 additional hours, and are completely dissipated by the end of the sixth hour. Qualitatively there were few visual effects reported with para-DOT (87), but in other aspects many of the conceptual and interpretively disruptive aspects of LSD intoxication were induced. With closed eyes, there was an easy visualization of hypnogogic images, which, although not of voluntary origin as to subject matter, could be terminated at will.
There are two positional isomers of para-DOT: one has the sulfur in the 2-position of the aromatic ring (2-thiomethyl-4,5-dimethoxyphenylisopropylamine, ortho-DOT) and the other in the 3- (or 5-) position (5-thiomethyl-2,4-dimethoxyphenylisopropylamine, meta-DOT). Both compounds have been described chemically (Jacob et al., 1977) but neither has yet been reported as being psychotomimetic.
A number of homologs of para-DOT (87) have been synthesized, containing a variety of alkyl and aryl groups on the sulfur atom in the 4-position (Shulgin, 1977, unpublished data). The immediate ethyl homolog 4-thioethyl-2,5-dimethoxyphenylisopropylamine, 88) is about twice as potent as the S-methyl counterpart 87, but has similar chronological and qualitative properties. Threshold effects are apparent at 2 mg orally, and at effective dosages of about 5 mg there is the first appearance of central effects at approximately 0.5 hr following administration. Quantitatively the intoxicative state develops over the following 1.5 hr. The psychological syndrome is largely dissipated after 8 hr, but some residual physical stimulation can persist and has interfered with sleep in some subjects. The body kinetics and metabolic fate of (88) have not been studied.