Abstract
An improved method for palladium(II)-catalyzed Wacker oxidation of cyclic and internal olefins is described. Addition of perchloric, sulfuric, nitric, or tetrafluoroboric acid to a chloride free solution of the Pd(II) catalyst gives rate enhancements of up to a factor of 50. The oxidation of cyclohexene to cyclohexanone, which was previously reported to give a 97 % yield after 5 h, is now accomplished in 1 h quantitatively, with only one-third of the amount of Pd(II) used. Limitations of the method are also discussed.
- RCH=CH2 + Pd2+ + H2O → RCOCH3 + Pd0
- Pd0 + [Ox] → Pd2+ + [Red]
The palladium(II) oxidation of terminal olefins to give methyl ketones (Wacker oxidation) is well established both as an industrial and an organic synthetic process (Eq. 1).2,3 Chemical oxidants (Ox) such as Cu(II), Fe(III), MnO2, heteropolyacids, or quinones are used to regenerate Pd(II) from Pd(0) (Eq. 2)4-10, thus making the reactions catalytic with respect to the Pd(II). Recently, electrochemical regeneration of Pd(II) in these systems has been reported11-13.
One of the deficiencies of this reaction is that the oxidation of cyclic and internal olefins is inefficient3. On the other hand, it is clear from early industrial work7,14-16 and the patent literature17,18 that the homogeneous oxidation of ethylene and terminal olefins is accelerated by addition of small amounts of acid but is inhibited by acid at high concentrations and by chloride7,14-16,19. However, the influence of acid on cyclic olefins is less certain. Moiseev et al.20 indicated that the cyclohexene oxidation rate was independent of [H+], whereas Kolb et al.8 and Horowitz12 demonstrate that the cyclic olefin oxidation rate is enhanced in the presence of inorganic acids. It does not appear, to our knowledge, that the rate enhancement gained by the addition of acids in a chloride-free system has been exploited in the laboratory-scale synthesis of ketones from terminal, internal, and cyclic olefins.
In this paper we report a general laboratory-scale method for the rapid, quantitative conversion of olefins to ketones. In this method inorganic acids containing non-complexing (i.e. weakly nucleophilic) anions are added to a Pd(II)/benzoquinone olefin oxidation system. We also have examined the performance of several Pd(II) salts in the presence of these acids and determined the optimum acid concentration range for each.
Results and Discussion
As part of ongoing studies with electrode-mediated Pd(II) oxidation reactions13 we found that it was necessary to use a well-established homogeneous reaction in order to optimize our electrochemical cell design. The Wacker oxidation of olefins seemed to be ideal for such studies. However, these reactions were generally too slow for electrocatalytic applications, and we found that it was necessary to try to improve the rates under the conditions used for the electrochemistry.
Table 1
The Effect of Acid of the
Wacker Oxidation of 1-Decene
| Acid (conv.)a | Products | Yield |
| HCl (0%) | no reaction | - |
| H2SO4 (93%) | 2-decanone | 64% |
| 3-decanone | 5% |
|
| 4-decanone | 14% |
|
(90%)b |
||
| HNO3 (93%) | 2-decanone | 83% |
| 3-decanone | 6% |
|
| decanal | 4% |
|
(100%)b |
||
| HClO4 (98%) | 2-decanone | 79% |
| 3-decanone | 10% |
|
| decanal | 4% |
|
(95%)b |
||
| HBF4 (99%) | 2-decanone | 84% |
| 3-decanone | 9% |
|
| decanal | 3% |
|
(97%)b |
||
Notes:
a) Conversion of 1-decene
after 10-min reaction at
60°C using 0.25 M acid.
b) Material balance based
on starting material.
The addition of strong acids to a solution of acetonitrile/water (7:1 v/v) containing 1-decene (0.2 M), Pd(OAc)2 (0.004 M), and benzoquinone (0.18 M) accelerated the Wacker oxidation by as much as a factor of 50 compared to a standard literature procedure3. The reaction was inhibited by HCl but accelerated by sulfuric, nitric, perchloric, or tetrafluoroboric acid (Table 1). In the latter two cases the conversion after only 10 min was quantitative and gave a higher selectivity toward 2-decanone, presumably because the olefin isomerization could not compete as effectively with oxidation in these cases. In principle, any strong inorganic acid possessing noncomplexing anions can be used to effect the acid acceleration.
The oxidation of a number of olefins has been examined using the reaction conditions described below. We found, surprisingly, that internal and cyclic olefins were rapidly (within 10 min) and quantitatively converted to their corresponding ketones (Table 2). This higher reactivity toward cyclic and internal olefins, which characteristically have been very difficult to oxidize with Pd(II)2-4, is a significant improvement.
Cyclohexene, 1-decene, and trans-2-octene were oxidized at 23°C as well as at 60°C (2 mol% catalyst). Complete conversion was obtained at both temperatures in all cases. The product distributions in the 1-decene oxidations at both temperatures were similar. However, the product distributions in the trans-2-octene reactions were temperature dependent with the 2-octanone/3-octanone ratio decreasing from 2.0 at 23°C to 0.85 at 60°C. In general, methyl ketones are the favored products in the oxidation of 2-alkenes8. However, olefin isomerization by Pd(II) is enhanced at higher temperatures21, leading to an increase in the formation of 3- and 4-ketones. Cyclohexanone was the only product formed in cyclohexene oxidations.
Table 2
Product Analysis from Pd(II) Alkene Oxidationa
| Substrate (conv.) | Temp | Productsb | Yieldc |
| 1-Octene (99%) |
60°C |
2-octanone |
82% |
| 3-octanone |
8% |
||
| 4-octanone |
2% |
||
| (93%)c |
|||
| 1-Decene (98%) |
60°C |
2-decanone |
79% |
| 3-decanone |
10% |
||
| decanal |
4% |
||
| (95%)c |
|||
| 1-Decene (94%) |
23°C |
2-decanone |
81% |
| 3-decanone |
2% |
||
| decanal |
3% |
||
| (92%)c |
|||
| trans-2-Octene (100%) |
60°C |
2-octanone |
40% |
| 3-octanone |
47% |
||
| 4-octanone |
11% |
||
| (98%)c |
|||
| trans-2-Octene (100%) |
23°C |
2-octanone |
53% |
| 3-octanone |
26% |
||
| 4-octanone |
5% |
||
| (86%)c |
|||
| cis-2-Heptene (100%) |
60°C |
2-heptanone |
58% |
| 3-heptanone |
32% |
||
| 4-heptanone |
7% |
||
| (97%)c |
|||
| Cyclohexene (100%) |
60°C |
cyclohexanone |
102% |
| (102%)c |
|||
| Cyclohexene (100%) |
23°C |
cyclohexanone |
102% |
| (102%)c |
|||
| Cycloheptene (92%) |
60°C |
cycloheptanone |
76% |
| (84%)c |
|||
| Styrene (98%) |
60°C |
acetophenone |
54% |
| 2-phenylethanal |
12% |
||
| (68%)c |
Notes:
a) Using 2 mol% Pd(OAc)2, 90 mol% benzoquinone
and 0.24 M perchloric acid.
b) Products identified by GC/MS.
Determined by GC using and internal standard (±2%).
c) Material balance based on starting material.
Selected examples from the literature, which are among the best results available, are compared with the results for 1-decene and cyclohexene oxidations using the method described in this paper (Table 3). Ogawa and co-workers5,6 reported the oxidation of cyclohexene using a PdSO4-heteropolyacid (H3PMo6W6O40) catalyst system.
In that work a turnover number of 85 (85% conversion) was obtained after 24-h reaction with 1 mol% PdSO4. Using the method described in this work a turnover number of 56 (56% conversion) was obtained after only 10 min, and complete conversion (100 turnovers) in 1 h. As in the oxidation of 1-decene, our results represent an improvement by a factor of ca. 50 in cyclohexene oxidation.
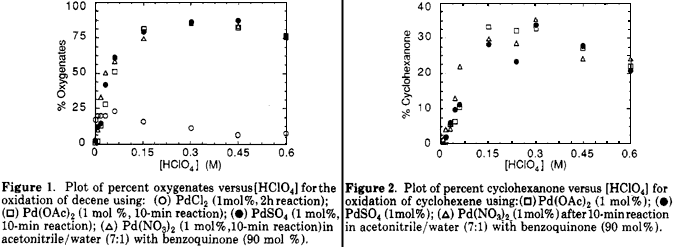
Ogawa noted6, using a heteropolyacid and O2 as the Pd(0) reoxidant, that palladium acetate exhibited a lower reactivity than the sulfate or nitrate salt (PdCl2 was the least reactive catalyst). While the role of the counterion does not appear to be well understood, there was a possibility that the use of these other palladium salts would enhance the reactivity of the catalyst toward cyclohexene. Concurrently, we wished to determine the optimum acid concentrations required for the olefin oxidations. Figures 1 and 2 show the acid concentration dependence in 1-decene and cyclohexene oxidations with Pd(OAc)2, PdSO4, Pd(NO3)2, and PdCl2 catalysts.
For the oxidation of 1-decene in the absence of acid only 2% conversion of the decene was obtained with all four palladium salts. There was essentially no difference among the acetate, sulfate, and nitrate catalysts in the reactivity as a function of perchloric acid concentration. The oxidation reaction was accelerated at low acid concentrations (up to 0.15 M) with a maximum between 0.30 and 0.45 M. At higher acid concentrations (>0.45 M) some inhibition was observed. Conversion levels attained in the palladium acetate, nitrate, and sulfate catalyzed reaction at acid concentrations above 0.01 M were significantly greater (by at least a factor of 5) than those obtained in any of the PdCl2-catalyzed 1-decene oxidations (in Figure 1 the points for PdCl2 were obtained after 2 h of reaction, rather than 10 min, in order to improve the precision of the GC measurements). The evidence presented above strongly supports a mechanism in which a common species participates in catalytic olefin oxidations involving Pd(II) salts possessing noncomplexing anions. The catalytically active species may have benzoquinone22, acetonitrile23, and/or aquo ligands coordinated to Pd(II), as well as the olefin. These data cannot be used to distinguish between the various possibilities.
The acceleration/inhibition effects induced by the addition of acid have been reported for a number of olefins7,8,12,19,24. Acceleration by acid may arise from protonation of the ligands coordinated to the Pd(II) which, in turn, increases the electrophilicity of Pd(II) and renders the Pd(II) more reactive toward the olefin23-25. The acid also may remove the noncomplexing anion thereby exposing a "bare" reactive Pd(II) atom or increase the rate of ligand exchange. In either case the result of acid addition is to increase the electron affinity of the central Pd(II) atom.
Originally it was proposed that the step involving formation of the cis-hydroxo-Pd-olefin complex was responsible for the acid inhibition in the Wacker oxidation7. However, work by Bäckvall26 and Stille27 demonstrated that the cis-hydroxo-Pd-olefin complex was not directly involved in the olefin oxidation. The currently accepted mechanism25-27 involves reversible nucleophilic attack on the coordinated olefin by the solvent. A proton is lost from the solvent molecule during the attack on the coordinated olefin, hence, at high proton concentrations the reactivity is suppressed.
Acceleration of the cyclohexene oxidation reaction using the acetate, sulfate, or nitrate salts was also observed at low acid concentrations (Figure 2), with the optimum conversion attained with an acid concentration of 0.30 M. Even at relatively low acid concentrations (0.03 M) the oxidation of cyclohexene is slower than 1-decene by only a factor of ca. 6. Cyclohexene was not oxidized by PdCl2. As in the case of 1-decene, there was inhibition of the reaction at higher acid concentrations. These results are consistent with those of Kolb et al.8 and Horowitz12, where acceleration and inhibition is observed in cyclic olefin oxidations.
The rate of oxidation of cyclohexanone was less reproducible than in the oxidation of 1-decene as evidenced by the larger scatter in the points in Figure 2. However, the trend is clear and, as in the case of 1-decene, the evidence favors a mechanism with a common catalyst for the reactions of palladium acetate, sulfate, and nitrate.
Table 3
| Substrate | Catalyst (mol%) | Reoxidant | Solvent | Temp | Time | Pd(II) turnovera |
| 1-deceneb | Pd(OAc)2 (1.0) | benzoquinone | MeCN/H2O | 23°C |
10 min |
77 |
| 1-deceneb | Pd(OAc)2 (2.0) | benzoquinone | MeCN/H2O | 23°C |
10 min |
43 |
| 1-deceneb | PdCl2 (1.0) | benzoquinone | MeCN/H2O | 23°C |
120 min |
17 |
| 1-decenec | PdCl2 (1.0) | benzoquinone | DMF/H2O | 30°C |
420 min |
78 |
| cyclohexeneb | Pd(OAc)2 (1.0) | benzoquinone | MeCN/H2O | 23°C |
10 min |
56 |
| cyclohexeneb | Pd(OAc)2 (1.0) | benzoquinone | MeCN/H2O | 23°C |
60 min |
100 |
| cyclohexened | PdSO4 (1.0) | H3PMo6W6O40/O2 | NMF/H2O | 30°C |
1440 min |
85 |
| cyclohexened | PdSO4 (3.3) | H3PMo6W6O40/O2 | NMF/H2O | 30°C |
300 min |
29 |
Notes:
a) Based on moles of olefin converted per mole of Pd(II).
NMF = N-methylformamide; DMF = dimethylformamide; MeCN = acetonitrile.
b) This work.
c) From Tsuji3.
d) From Ogawa et al.5
Conclusions
We have found that the addition of moderate to large amounts inorganic acids to the Pd(II)/benzoquinone oxidation of terminal, internal, and cyclic olefins in a chloride-free reaction leads to rate enhancements of up to a factor of 50 over those reported in the literature. Even at low acid concentrations (0.01 M) rate enhancements of up to a factor of 5-10 over the traditional PdCl2 reactions are obtained. There are still some limitations to this method. Trisubstituted olefins (both cyclic and acyclic) as well as conjugated dienes are not oxidized by the method described. Furthermore, compounds which undergo facile acid-catalyzed reactions also may not be amenable to this approach. However, this method will be of value for synthetic applications involving internal and cyclic olefins where, until now, the Pd(II)-catalyzed oxidation was not a viable alternative.
Experimental
All solvents, olefins and palladium catalysts were commercially available (Aldrich) and were used as received. Benzoquinone was filtered through a silica gel column (eluting with CHCl3) and recrystallized twice from hexane. All reactions were carried out using a procedure similar to the one described below.
Acid Dependence
Palladium(II) acetate (0.2 mmol), benzoquinone (9 mmol), and the inorganic acid (HCl, HClO4, HBF4, H2SO4, or HNO3, 0.1 M) were dissolved in acetonitrile/water (7:1 v/v, 50 mL). The solution was deoxygenated by purging with argon for at least 30 min and stirred vigorously until the Pd(OAc)2 had dissolved. The olefin (10 mmol) was then added to the flask (by syringe), and the reaction mixture was stirred for 10 min. The products were separated from the catalyst by extraction into hexane or diethyl ether, washed with 30% aqueous sodium hydroxide, and then analyzed by a capillary GC-internal standard method. n-Decane was the internal standard in cyclohexene and cycloheptene reactions, and both n-tridecane and n-hexadecane were used as internal standards in all other reactions.
Olefin Oxidation Reactions
Oxidations of various olefins listed in Table II were performed in the manner described above. In this case, perchloric acid (72% w/v, 1.0 mL) was used, and the reactions were carried out at either 23°C or 60°C as noted in Table II. The cyclohexene and 1-decene reactions listed in Table III were also performed in the method similar to that described above. For cyclohexene, the total volume was only 20 mL.
Perchloric Acid Concentration Dependence
In all reactions 0.1 mmol of the Pd(II) salt, 10 mmol of the olefin, 9 mmol of benzoquinone, and varying amounts of HClO4 (72% w/v) in a total volume of 20 mL were used. The reaction time was 10 min. The reaction mixture was then extracted into diethyl ether, washed with 30% (w/v) aqueous NaOH, and finally dried over MgSO4. Products were analyzed by a capillary GC-internal standard method, with both n-tridecane and n-hexadecane used as internal standard.