(Hive Bee)
07-21-03 02:17
No 448729
(Rated as: excellent)
Chimimanie's voice:
This paper describe the preparation of 5-Cyano-DMT, 5-Nitro-DMT and good ol straight DMT in a way i find somebees will love
They alpha-brominate 3-acetyl-indole, swap the Br with dimethylamine, then reduct the ketone to the alkane (not the alcohol!
For DMT, the yield is 49% in the reduction step, 71% for the swap step, and no yield is given for the bromination step, but it should bee more than 75%.
NB: I added some italics and some bold on part of text relevant to DMT.
And here the fun start:
The Preparation and Reduction of 5-Cyano-3-indolylketones. Synthesis of 5-Cyanotryptamines, Joseph I. DeGraw, Jill G. Kennedy and W. A. Skinner, J. Het. Chem. 1966, p9.
Abstract:
The acylation of indoles under acidic conditions has been studied. Stannic chloride was shown to be an effective catalyst for the preparation of some 3-acylindoles, notably 5-cyano-3-indolylketones. The various 5-cyano-3-indolylketones were reduced with sodium borohydride to yield either the 5-cyano-3-carbinols or 5-cyano-3-alkylindoles. 5-Cyanotryptamines were obtained by reduction of appropriate alpha-dialkylamino and alpha-azidoketones. A cleavage reaction of the carbinols involving loss of the 3-side chain to yield 5-cyanoindole is also described.
The usual method for the preparation of 3-indolylketones has been the reaction of an indolylmagnesium halide with an appropriate acid chloride. However, this method cannot be employed in the presence of functional groups that are incompatible with Grignard reagents. The synthesis of 3-indolylketones by acylation under acid catalyzed conditions has not been extensively investigated. In a recent publication, which indicated 3-acylindoles to be of value as antidepressants, Szmuszkovicz, et al. [1] prepared some of these ketones by the action of phosphoryl chloride and N,N-dimethyl carboxamides on indole. Anthony [2] had previously investigated this method of preparation even more extensively, obtaining good yields in most cases studied. The synthesis of 5-nitro-3-acetylindole by the stannic chloride catalyzed reaction of 5-nitroindole and acetic anhydride has also been reported [3]. In this work we have extended the use of the stannic chloride procedure and made a comparison with some other methods, using 5-cyanoindole as a reactant.
The reaction of 5-cyanoindole with anhydrous stannic chloride and an acid chloride in benzene afforded the 5-cyano-3-indolylketones in yields of 38 to 70%. Indole itself gave mostly red tars under these conditions. It was found that acid chlorides gave better yields and cleaner products in this reaction, than did the acid anhydrides. This may be due to the lower quantities of stannic chloride required to condense the acid chlorides. The effects of variation of the solvent or the nature of the "Lewis acid" were not investigated.
Three other general procedures involving acid catalyzed acylation were also studied. The first of these was the phosphoryl chloride-carboxamide method discussed above. When 5-cyanoindole was treated with phosphoryl chloride and N,N-dimethylacetamide at 85-90° according to the procedure of Anthony [2], only unchanged 5-cyanoindole was obtained. This seems remarkable in view of the facile acylations of indole obtained by this method. 5-Cyano-3-indolylaldehyde (I) was readily prepared, however, by the phosphoryl chloride and dimethyl formamide method [4]. The second method investigated involved the use of polyphosphoric acid [5] and acetic acid at 55° which gave only a 17% yield of 3-acetyl-5-cyanoindole (II). A third and more successful process was the trifluoroacetic anhydride [6] catalyzed reaction of acetic acid with 5-cyanoindole. When the reactants were used in a 2:1:6:1 molar ratio, respectively, a 68% yield of 3-acetyl-5-cyanoindole was obtained. The treatment of 5-cyanoindole with trifluoroacetic anhydride alone gave an 88% yield of the 3-trifluoroacetyl derivative (VII).
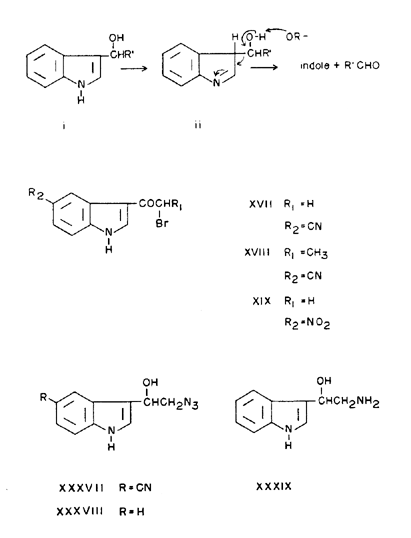
Reduction of ketones (III, IV, VI, VII) with sodium borohydride in hot ethanol yielded the corresponding alcohols as crystalline solids. However, the methylketone (II), under a variety of conditions, would only afford an unstable syrup, free of carbonyl absorption in the infrared. The phenylketone (V) gave an impure, carbonyl-free product that was mostly 3-benzyl-5-cyanoindole. Reduction of V at lower temperatures gave a product which was mainly the alcohol, but analytically impure.
Prolonged treatment of the ketones with sodium borohydride in refluxing 1-propanol gave unexpected results. The main products of the reductions were the corresponding 3-alkyl-5-cyanoindoles. This in itself was not unusual since Ames, et al. [7] reduced 3-acetylindole to 3-ethylindole with lithium borohydride in hot tetrahydrofuran. However, an accompanying side product, which was formed in significant amounts in each case, was found to be 5-cyanoindole. This indicates that a side chain fragmentation occurs in competition with the reduction [8]. A plausible mechanism for the loss of the 3-substituent during the borohydride reduction of 3-acylindoles is the following. The ketone undoubtedly is first reduced to the hydroxy compound (i) which then undergoes a base catalyzed decomposition requiring an isomerization of the indole system to an indolenine. The indolenine intermediate (ii) would then suffer exchange of the alcohol proton to the alkoxide medium and a collapse of the resulting ion to form an aldehyde and indole. The aldehyde fragment would of course be reduced to the corresponding alcohol. To partially support this mechanism the 5-cyano-3-(1-hydroxy-2-phenethyl)indole (X) was treated with sodium borohydride in 1-propanol and yielded 5-cyanoindole and 5-cyano-3-phenethylindole (XVI).
A more useful extension of this work was its application to the synthesis of 5-cyanotryptamines by the borohydride reduction of appropriate 5-cyano-3-dialkylaminomethyl- and 5-cyano-3-azidomethylindolylketones. These tryptamines would not be obtainable from other methods requiring the use of powerful reducing media.
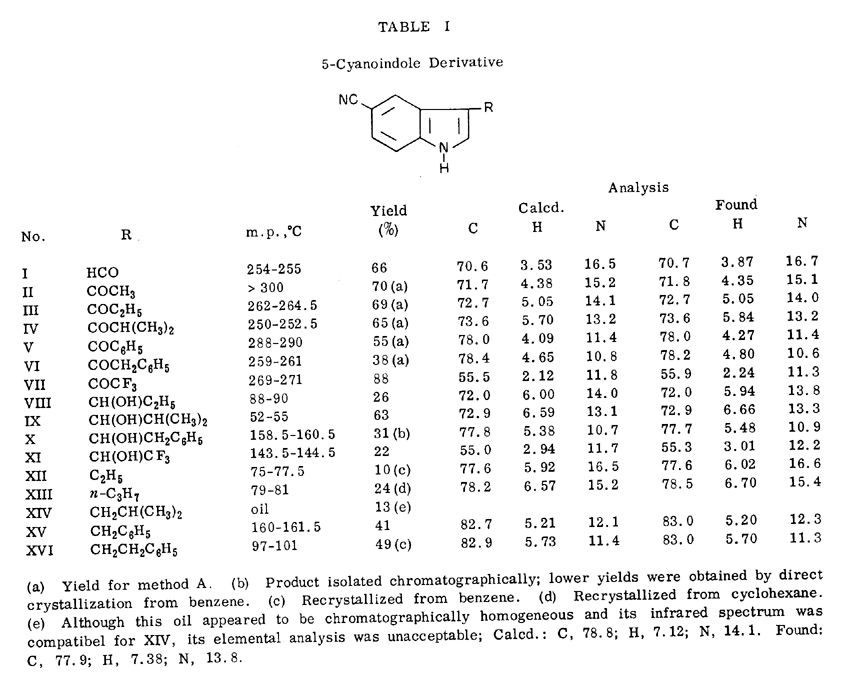
3-Acetyl-5-cyanoindole (II) was treated with bromine in methanol-dimethylformamide and the resulting alpha-bromoketone (XVII) was allowed to react with various secondary amines to yield the 5-cyano-3-dialkylaminomethylketones (XXI-XXIV), essentially following the procedure of Bodendorf and Walk [9]. The azido ketone (XX) was readily prepared by displacement of the bromine atom with sodium azide in aqueous dimethylformamide. Bromination of 5-cyano-3-propionylindole (III) could not be carried out by the above method, but was achieved by the novel reagent, trimethylphenylammonium tribromide [10] to yield the bromoketone (XVIII). Displacement of the bromine was likewise readily effected with dimethylamine or sodium azide.
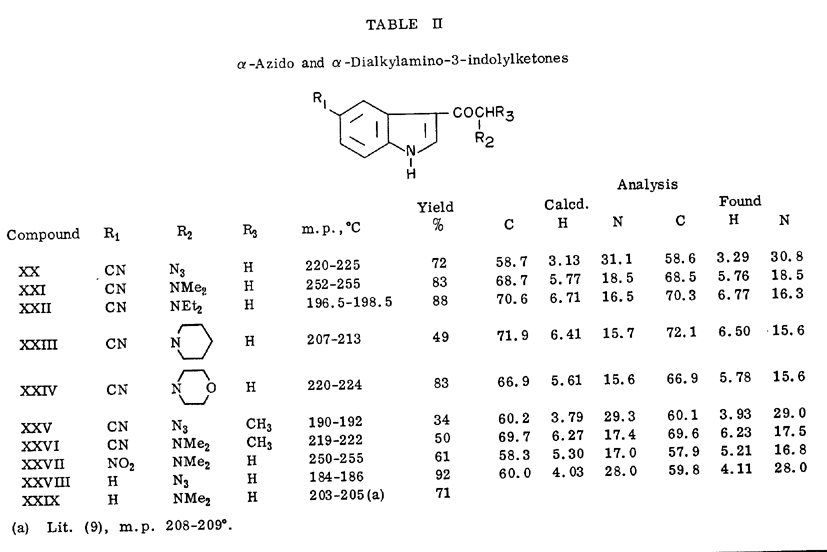
Reduction of the azido ketone (XX) or the dialkylamino ketones (XXI - XXIV) with sodium borohydride in refluxing 1-propanol afforded the corresponding substituted 5-cyanotryptamines (XXX - XXXIV) along with 5-cyanoindole. However, the 5-cyano-3-azido and dimethylaminopropionyl ketones (XXV and XXVI) afforded only meager yields of nonidentifiable products when reduced with sodium borohydride under a variety of conditions. In addition 5-nitro-N,N-dimethyltryptamine (XXXV) was prepared from the 5-nitro-3-dimethylaminoketone (XXVII) by reduction in 1-propanol. The latter tryptamine was previously synthesized by Shaw and Wooley [11] from 5-nitro-3-chloroethylindole.
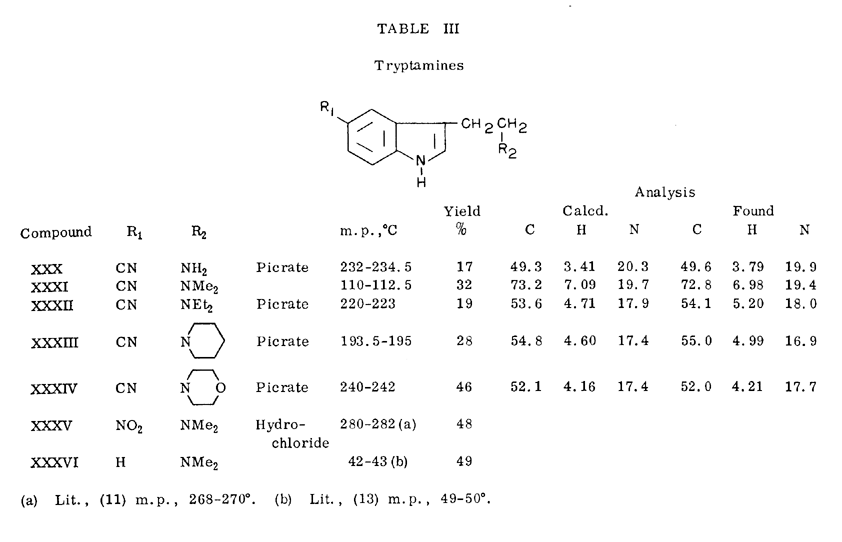
Attempts to prepare the hydroxyamines from the aminoketones by reduction with sodium borohydride under milder conditions [12] were unsuccessful. The 5-cyano-3-dialkylaminomethylketones were fairly resistant to reduction with sodium borohydride in methanol or ethanol. In contrast, even the hindered 5-cyano-3-acylindoles were easily reduced to alcohols in those solvents. The 5-cyano-3-azido ketone (XX) underwent facile reduction to the hydroxyazide (XXXVII) in ethanol at room temperature.
It was also of interest to examine the behavior of compounds related to those previously discussed, but without substituents on the indole nucleus. Reduction of the dimethylaminoketone derivative (XXIX) [7,9,12] with sodium borohydride in hot 1-propanol gave N,N-dimethyltryptamine (XXXVI) and indole as expected. 3-Azidoacetylindole (XXVIII) could be easily reduced with borohydride to the hydroxy azide (XXXVIII). Hydrogenation of the hydroxyazide over platinum oxide afforded 3-(2-amino-1-hydroxyethyl) indole (XXXIX), isolated as its picrate salt [7].
(Hive Bee)
07-21-03 02:18
No 448730
(Rated as: excellent)
Experimental:
Reaction of 5-Cyanoindole with:
A. Acid Chlorides and Stannic Chloride.
The following preparation of 3-acetyl-5-cyanoindole (II) is representative of the procedure used to prepare the related ketones listed in Table I. To an ice cold suspension of 25.0 g. (0.176 mole) of 5-cyanoindole in 400 ml. of benzene containing 21.5 ml. (0.302 mole) of acetyl chloride was added, dropwise with stirring, a solution of 35.5 ml. (0.305 mole) of anhydrous stannic chloride in 100 ml. of benzene. An orange-red complex precipitated and the mixture was stirred at 0-5° for 1 hour and mixed with 1250 ml. of ice cold water. The resulting mixture was stirred for 30 minutes at 0-5° and the solid collected by filtration, washed thoroughly with water and dried. The crude material was treated with 800 ml. of hot acetone and allowed to stand for a few hours. The product was collected and dried to yield 20.0 g. of off-white crystals, m. p.>300°. A second crop of 2.8 g., m. p. >300°, was obtained by concentration of the mother liquors to 300 ml. The total yield of 3-acetyl-5-cyanoindole was 22.8 g. or 70%.
B. Acid Anhydride and Stannic Chloride.
This reaction was conducted as above with 3.90 g. (27 mmoles) of 5-cyanoindole, 4.3 ml. (43 mmoles) of acetic anhydride and 9.7 ml. (80 mmoles) of stannic chloride to give a crude yield of 4.67 g. (93%), of II, m. p. 230-300°. This material was dark colored and required a recrystallization from 75 ml. of acetonitrile to yield 3.26 g. (65%), m.p. 300-305°. The purified material was still of inferior quality as compared with that obtained by the acid chloride procedure. Runs with propionic anhydride gave similar results.
C. Trifluoroacetic Anhydride.
To 12 ml. of ice cold trifluoroacetic anhydride was added slowly 2.00 g. of 5-cyanoindole and the mixture was stirred at room temperature for 4 hours. The mixture was evaporated to dryness in vacuo and the residue was stirred between 10 ml. of ether and 10 ml. of saturated aqueous sodium bicarbonate solution. The solid was collected, washed thoroughly with water and ether and dried to leave 2.91 g. (88%) of the trifluoroketone (VII). An analytical sample was obtained by recrystallization from ethanol.
Reduction of Ketones with:
A. Sodium borohydride in Ethanol.
Ethanolic solutions of equal weights of the ketones and sodium borohydride were refluxed for 1 hour. The solvents were removed in vacuo and the residues diluted with water. Solid products at this stage were collected by filtration and liquid ones extracted into chloroform. The crude products were either extracted with hot benzene and recrystallized from the solvent or were chromatographed on silica gel. The methyl ketone (II) gave only an unstable syrup when reduced at 0-5°, room temperature or at reflux. Its infrared spectrum showed no carbonyl absorption at 6.10u. The phenyl ketone (V) gave mostly 3-benzyl-5-cyanoindole (XV) when run at reflux as shown by infrared comparison with analytically pure material. When the reaction was conducted at room temperature a crystalline solid, m.p. 129-131.5°, was obtained whose analysis indicated it to be mostly the alcohol.
B. Sodium Borohydride in 1-Propanol.
Solutions of the ketones (1 g.) and sodium borohydride (2 g.) in 2,55 ml. of 1-propanol were stirred at reflux for 15 hours. The solvent was evaporated in vacuo and the residues were partitioned between chloroform and water. Evaporation of the chloroform gave 70-90% yields of crude products, which showed the presence of two spots (Rf, 0.5 and 0.6-0.75) on silica gel thin layer plates when developed with chloroform and detected with iodine vapor. The spot at Rf, 0.5 was identical with that of 5-cyanoindole and the faster moving spots were the 3-alkyl-5-cyanoindoles (XII-XVI). No spots corresponding to the previously described ketones or alcohols (Rf, 0-0.2) were detected. The alkylindoles were obtained in low yield either by repeated recrystallization from benzene or cyclohexane or by column chromatography on silica gel. From the reduction of the isobutylketone (IV), 5-cyanoindole was isolated by column chromatography and identified by its infrared spectrum. The trifluoroketone (VII) was not investigated under these reduction conditions. Exposure of the alcohol (X) to the above reduction conditions afforded 5-cyanoindole and 5-cyano-3-phenethylindole (XVI) as shown by thin layer chromatography.
3-Bromoacetyl-5-cyanoindole (XVII).
To a solution of 5.00 g. (27.2 mmoles) of 3-acetyl-5-cyanoindole (II) in 50 ml. of dimethylformamide at 50° was added slowly a solution of 2.5 ml. (45.8 mmoles) of bromine in 75 ml. of methanol. The mixture was stirred for one hour at 50° and allowed to stand at room temperature for 15 hours. The white precipitate that formed was collected, washed with water, and dried to leave 5.45 g. (76%) of product, m. p. 278-284° (dec.). An analytical sample, m. p. 288-292°, was obtained by recrystallization from acetonitrile.
3-(2-Bromopropionyl)-5-cyanoindole (XVIII).
A mixture of 2.01 g. (10.3 mmoles) of 5-cyano-3-propionylindole (III), 3.6 g. (9.6 mmoles) of trimethylphenylammonium tribromide (10) and 75 ml. of tetrahydrofuran was stirred at 35-40° for 27 hours. The solvent was removed in vacuo and the residue stirred with 100 ml. of water. The insoluble product was collected, washed thoroughly with water, and dried to leave 2.81 g. (99%), of XVIII, M.P. 259-262° (dec.). Recrystallization from acetone of material from another run provided an analytical sample, m.p. 244-246°. Considerable variation in melting points was observed for different runs, although the infrared spectra were essentially the same.
3-Bromoacetyl-5-nitroindole (XIX).
This bromoketone was prepared by the same procedure for 3-bromoacetyl-5-cyanoindole (XVII). The product, m.p. >300°, was obtained in 46% yield after recrystallization from ethanol-dimethylformamide.
alpha-Azido-3-indolylketones.
A mixture of the appropriate 5-cyano-alpha-bromoketone (1 g.), sodium azide (2 g.) and 83% dimethylformamide (60 ml.) was stirred at 40° for 15 hours. The mixture was diluted with 30 ml. of water and the precipitate was collected, washed with water and dried. The crude products were recrystallized from aqueous dimethylformamide. 3-azidoacetylindole (XXVIII) was similarly prepared from 3-bromoacetylindole [9] but in 83% methanol and the product was recrystallized from ethanol. The azido ketones exhibited prominent bands at 4.7 microns in the infrared. The data for these compounds appear in Table II.
alpha-Dialkylamino-3-indolylketones.
The various alpha-bromoketones were allowed to react with a 3-4 mole excess of an appropriate secondary amine in refluxing 2-propanol, according to the general procedure of Bodendorf and Walk [9]. The data for these compounds are presented in Table II.
Substituted Tryptamines.
The appropriate dialkylaminoketone or azidoketone was refluxed with twice its weight of sodium borohydride in 1-propanol for 15 hours. The solvent was evaporated in vacuo and the residue was partitioned between water and chloroform. The chloroform extract was extracted with 3N-hydrochloric acid to separate the tryptamines from accompanying 3-unsubstituted indoles. 5-Cyanoindole (identified by infrared analysis and mixed melting point determination) was obtained from the reduction of XXI and indole (identified by infrared spectrum) was obtained from XXIX. The acid extracts were alkalized with 10% sodium hydroxide and the liberated bases extracted into chloroform. The tryptamines were isolated either as the free base or picrate salt. The data for the tryptamines appear in Table III.
3-(2-Azido-1-hydroxyethyl)-5-cyanoindole
A mixture of 0.50 g. (13.3 mmoles) of sodium borohydride, 0.50 g, (2.2 mmoles) of 3-azidoacetyl-5-cyanoindole (XX) and 10 ml. of ethanol was stirred at room temperature for one hour. The ethanol was evaporated in vacuo and the residue was taken up in 30 ml. of water. The aqueous solution was extracted with four 15-ml. portions of chloroform. The chloroform extract was dried over magnesium sulfate and evaporated in vacuo to leave 0.43 g. (84%) of a syrup, which later solidified to give pale green plates, m. p. 121-123°
3-(2-Azido-1-hydroxyethyl)indole (XXXVIII).
To a stirred suspension of 0.70 g. of 3-azidoacetylindole (XXVIII) in 28 ml. of methanol was slowly added 0,70 g, of sodium borohydride. The mixture was stirred at ambient temperature for 1 hour and evaporated in vacuo. The residue was partitioned between 30 ml. of water and 30 ml. of ether. The ether extract was washed with 10 ml. of water, dried over magnesium sulfate and evaporated in vacuo to leave 0.53 g. of a clear syrup
3-(2-Amino-1-hydroxyethyl)indole Picrate (XXXIX).
A mixture of 0.53 g. of the azido alcohol (XXXVIII), 50 mg. of platinum oxide and 10 ml. of ethanol was stirred under an atmosphere of hydrogen for 5 hours. Only uptake for the catalyst was observed. The catalyst was removed by filtration and the filtrate was evaporated in vacuo to leave a viscous syrup. The infrared spectrum showed complete loss of the azide band and stronq OH, NH absorption with association typical of amino alcohols. The syrup was dissolved in 5 ml. of ethanol and added to 0.65 g. of picric acid in 50 ml. of warm water. The resulting yellow, crystalline precipitate was collected, washed with water and dried to leave 0.80 g., m.p. 146-150° (dec. ). A 200 mg. portion was recrystallized from 5 ml. of ethanol to yield 55 mg. of picrate, m.p. 148-150° (dec. ). Ames, et al.[7] had previously obtained this material by reduction of 3-N-carbobenzoxy-glycylindole; picrate, m.p. 100° (dec.).
References:
[1] . H. H. Keasling, R. E. Willette and J. Szmuszkovicz, J. Med. Chem., 7, 94 (1964).
[2] . W. C. Anthony, J. Org. Chem., 25, 2049 (1960).
[3] . J. DeGraw and L. Goodman, J. Med, Chem., 7, 213 (1964).
[4] . G. F. Smith, J. Chem, Soc., 3842 (1954).
[5] . P. D. Gardner, J. Am. Chem. Soc., 76, 4550 (1954).
[6] . E. Bourne, M. Stacey, J. Tatlow and J. Tedder, J. Chem. Soc., 718 (1951).
[7] . D. E. Ames, R. E. Bowman, D. D. Evans and W. A. Jones, ibid., 1984 (1956).
[8] . Since the completion of this work it was shown by J. C. Powers (Tetrahedron Letters, 655, (1965)) that a variety of 3-substituted indoles can be cleaved by alkali. He has also suggested a mechanism involving an indolenlne intermediate.
[9] . K. Bodendorf and A. Walk, Arch. Pharm., 294, 486 (1961).
[10]. A. Marquet and J. Jacques, Bull. Soc, Chim. France, 90 (1962).
[11]. E. Shaw and D. W. Wolley, J. Am. Chem. Soc., 75, 1877 (1953).
[12]. W. C. Anthony and J. Szmuszkovicz, Patent US2821532.
[13]. T. Hoshino and K. Shimodaira, Ann., 520, 19 (1935).
(Hive Bee)
07-21-03 16:27
No 448820
(Rated as: excellent)
Organic Letters (2001), 3(7), 1005-1007 (http://www.angelfire.lycos.com/scifi2/l
DOI:10.1021/ol007056i
Acylation of Indole under Friedel-Crafts Conditions
An Improved Method To Obtain 3-Acylindoles Regioselectively
Abstract
The reaction of unsubstituted indole with different acylating agents such as acid chlorides, anhydrides, nitriles, and amino acid derivatives in the presence of Lewis acid is reported.
Table 1. Effectiveness of Different Lewis Acids in Obtaining 3-Acylindole
| Entry | Acylating agentb (AA) | Lewis acid (LA) | ratio indole/AA/LA | reaction time/ha | product (R) | Yields % |
| 1 | Acetanhydride | AlCl3 | 1/1/2 | 10 | CH3 | 78 |
| 2 | Acetanhydride | TiCl4 | 1/1/2 | 10 | CH3 | 50 |
| 3 | Acetanhydride | SnCl4 | 1/1/2 | 4 | CH3 | 42 |
| 4 | CH3CN | SnCl4 | 1/10/1.2 | 4 | CH3 | 96 |
| 5 | Acetyl chloride | AlCl3 | 1/1/1.2 | 6 | CH3 | 71 |
| 6 | Acetyl chloride | SnCl4 | 1/1/1.2 | 2 | CH3 | 95 |
| 7 | Propionyl chloride | SnCl4 | 1/1/1.2 | 10 | CH2CH3 | 80 |
| 8 | N-Acetylchloride benzylamine | SnCl4 | 1/1/1.2 | 10 | CH2NHCH2Ph | 40 |
| 9 | Succinic acid dichloride | SnCl4 | 1/1/4 | 10 | CH2CH2COOH | 72 |
| 10 | Chloro acetylchloride | SnCl4 | 1/1/1.2 | 8 | CH2Cl | 80 |
| 11 | Bromo acetylchloride | SnCl4 | 1/1/1.2 | 8 | CH2Br | 70 |
| 12 | Bromo acetylbromide | SnCl4 | 1/1/1.2 | 8 | CH2Br | 50 |
a Refers to the period of time after the complete addition of nitromethane.
b The acylating agent was slowly added to the reaction mixture at 0 °C.
3-Acetylindole.
General Procedure.
To a stirring solution of indole (1.17 g, 10 mmol) in CH2Cl2 (20 mL) under argon at 0 °C was added SnCl4 (1.44 mL, 12 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then acetic anhydride (10 mmol) was added in small portions to the suspension, followed by nitromethane (15 mL). The mixture was stirred for 2 h at room temperature. After being quenched with ice and water (30 mL), the mixture was filtered to remove inorganic precipitates, and the organic material was extracted with ethyl acetate (50 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to give the product as a crystalline solid.
Lego's voice: Thanks to Chimimanie this article is now useful to the Hive. Entry 10-12 give a similiar product like (XVII) in the above post. It is one step less from indole to DMT and other dialkyltryptamines.
The candle that burns twice as bright burns half as long
(Hive Bee)
07-21-03 17:36
No 448825
Thank you Lego
I really like the one with acetonitrile and tin chloride!
The one with bromoacetyl chloride is useful to, reducing the global route by one step...
So now it is:
0.70 * 0.71 * 0.49 = ~25% from indole to DMT in 3 steps, give or take
This is an useful route to tryptamines, and it sure benefit from your acylation post, thanx again dude
(Hive Bee)
10-14-04 16:51
No 535821
(Rated as: excellent)
3-Acylindoles via a One-Pot, Regioselective Friedel–Crafts Reaction
James H. Wynne, Christopher T. Lloyd, Samuel D. Jensen, Sean Boson, Wayne M. Stalick
Synthesis, 2004, 14, 2277-2282
DOI:10.1055/s-2004-831177
Abstract: Within we report a general one-pot high-yielding Friedel-Crafts acylation of indole using acid chlorides and diethylaluminum chloride in gram scale quantities. This general synthesis affords products that are easily isolated and require no complex purification procedures.
General Procedure: Into a 100 mL round-bottomed flask equipped with magnetic stir bar and a pressure equalizing addition funnel was placed indole (3.55 g, 20 mmol) and CH2Cl2 (50 mL). Nitrogen was used to copiously flush the apparatus for a few minutes through a syringe needle piercing the septum on the addition funnel. The solution was cooled to –78 °C in a dry ice–acetone bath for 15 min before the addition of Et2AlCl–hexanes (1 M; 20 mL) into the addition funnel via an 18 gauge cannula. Slow, dropwise addition of the Et2AlCl was made over a 10 min period while maintaining the –78 °C reaction temperature.
The solution turned yellow yet remained transparent. The bath was exchanged for a salt–ice bath and the solution armed to –10 °C. The resulting yellow solution was allowed to stir for 15 min before the slow dropwise addition of the corresponding acyl halide (30 mmol) was made employing the same addition funnel at a rate of 1 drop every 3 seconds. The solution changed to an orange color.
The solution was allowed to stir for 2 h during which time the reaction temperature gradually warmed to r.t. For some substrates, higher rates of magnetic stirring were required to ensure adequate mixing as a result of precipitate formation.
CAUTION Very slow addition of an aliquot (10 mL) of pH 7 buffer solution was made at a rate of 1 drop every 10 s. Extreme caution was taken to avoid the violent bubbling or foaming that ensues from this neutralization. It is believed that the bubbling is a result of ethane evolution from the neutralization of the aluminum catalyst.
Work-up A
The whitish suspended solid produced upon neutralization was vacuum filtered, washed with CH2Cl2 (50 mL) and allowed to dry. The dried solid was dissolved in acetone with minimum heat; sometimes below reflux, to avoid a color change from light pink to dark red. Any residual solid not dissolved in the acetone was filtered and discarded.
The acetone layer was allowed to concentrate to 50% of its volume without heating and then cooled in an ice bath before isolation by filtration to afford the pure desired crystalline product.
Work-up B
The aryl- and fluoro-containing 3-acylindoles were more soluble in CH2Cl2 than were the 3-alkyl acylindoles. In these cases, very little of the whitish suspended solid was produced upon neutralization of the reaction mixture. That which was formed was a small amount of the aluminum salts that were filtered and discarded to avoid emulsification in later steps of isolation. The filtered liquid contained most of the product in the organic layer and likewise most of the aluminum salts in the buffered aqueous layer. The filtrate was extracted with additional CH2Cl2 (2 × 35 mL). The aq layer containing the majority of the aluminum salts was discarded. The CH2Cl2 layer was washed with H2O (3 × 25 mL), dried (MgSO4), and evaporated to afford the crude product. The crude product was recrystallized from acetone using minimal heat as specified in work-up A.
Thanks to Java for retrieving this article!
The tendency is to push it as far as you can