07-06-03 07:49
No 444883
After getting frustrated from yet another 'tryptophan Cu-chelate in DMSO decarboxylation' failure (I just can't seem to isolate anything other than a black tarry mess
Then these posts came to mind again:
Post 122774 (dormouse: "Prodrugs -Lilienthal", Serious Chemistry)
Post 202791 (Lilienthal: "Re: N,N dimethyltrytOPHAN", Tryptamine Chemistry)
Seems like the final word is not yet said about the decomposition products obtained by pyrolysis of N,N-dimethyltryptophan
I would like to try a HCHO/NaBH4 methylation, but I'd be nice if we had some more journal reference backup, though.
Only related thing I've found so far is this:
J Chem Soc (1941) p 157
alpha-hydroxymethylamino-beta-3-indolylp
L-tryptophan (0.6 gr) was dissolved in water (12 ml), formalin (2 ml) was added, and the mixture was incubated at 38°C for not more than 3-4 hours. The colourless, crystalline but rather granular solid which had then separated was collected, washed with cold water, and dried in a vacuum. Yield: 60%
When the reaction mixture was incubated for 15 hours at 38°C in presence of NaOH, the beta-carboline was obtained in 80% yield.
http://www.poedecoder.com/Qrisse/works/d
(Hive Addict)
07-06-03 08:15
No 444888
can't help you on the dimethyltryptophan. but the Cu-chelate method
has me puzzled too. on addition of water there has always been this
sticky yellow precipitate, then on extraction with EtOAc => black tar.
bummer. but thanks a lot for reporting it.
(Pioneer Researcher)
07-06-03 11:58
No 444936
It was described here a decarboxylation in tryptophan by single heating in DMF. I tried it and it was an obvious evolution of CO2. I didn't isolate and verfy the priodcut, but it seemed to work acording to the external signals. It would be good if you try it and verify that the final prouct is tryptamine, the procedure is so easy and you can find it with the search engine.
(Hive Bee)
07-06-03 12:32
No 444947
Thanks, I've seen it and considered it.
I'm sure the Cu-chelate procedure produces tryptamine too, but isolating it is a whole different matter. Other things I've tried were: heating in DMSO, DMSO + various ketones, turpentine + d-carvone and even microwaving 5 gr tryptophan in 50 gr DMSO + 2 ml d-carvone for 6 min at 500W
I think will give up on these, maybe instead try the known tetralin, cyclohexanol or diphenylmethane routes.
I really want to synth N,N-dimethyltryptophan anyway. Maybe it's something, maybe not, doesn't matter much, at least we'll know then. So any hints or refs are much appreciated.
Should I just substitute tryptamine by 26mmol L-tryptophan in the HCHO/NaBH4 route ?
Post 431982 (Lego: "Article on DMT derivate synths", Tryptamine Chemistry)
http://www.poedecoder.com/Qrisse/works/d
(Moderator)
07-06-03 12:55
No 444954
Cool idea! Check my homepage for literature references:
http://www.fortunecity.com/westwood/stor
By using reductive alkylations you will be running into the same problems like with tryptamine alkylation: Pictet-Spengler ring closure.
Why not try the phenylhydrazine route? E.g. with 2-dimethylamino-5,5-dimethoxy-pentanoic acid methyl ester (e.g. from N,N-dimethyl-glycine ester and 3-halo-propionic aldehyde acetal).
(Hive Bee)
07-06-03 14:28
No 444988
By using reductive alkylations you will be running into the same problems like with tryptamine alkylation: Pictet-Spengler ring closure
Yes, but I wonder to what extent I'll occur? I still have to read up more on Pictet-Spengler, but I wonder why one substrate is more affected then another? (eg. tryptamine vs. phenylethylamine)
N,N-dimethyltryptophan benzylester from tryptophan benzylester with formaline, acetic acid, and NaCNBH4, yield 17% J. Med. Chem. 37 1269 (1994)
But then, on the other hand:
imines from tryptamine and 3-Meo-, 4-Meo- or 4-dimethylamino-benzaldehyde by precipitation after 90 min reflux in MeOH, Pictet-Spengler reaction to beta-carbolines with HCl, imine reductions with NaBH4 in EtOH, N-2-phenoxy- and N-benzyl-tryptamides and -amines
Coll. Czech. Chem. Commun. 28 629 (1963)
Why not try the phenylhydrazine route?
The gramine + ethyl dimethylamino-malonate route looks even more interesting to me. I can get Ann. Chim. but I guess Ann. Chim.(Rome) is a different journal?
http://www.poedecoder.com/Qrisse/works/d
(Hive Bee)
07-06-03 16:59
No 445024
DOI:10.1139/v02-083
Sodium borohydride: A versatile reagent in the reductive N-monoalkylation of a-amino acids and a-amino methyl esters
Giancarlo Verardo, Paola Geatti, Elena Pol, and Angelo G. Giumanini
Can. J. Chem./Rev. Can. Chim. 80(7): 779-788 (2002)
Full text (PDF 99 kb)
Abstract: a-Amino acids and a-amino methyl esters are easily converted to their N-monoalkyl derivatives by a reductive condensation reaction using several carbonyl compounds in the presence of sodium borohydride. This reducing agent has shown a wide versatility with minor but essential procedural variations. The reaction allows the a-monodeuterium labeling of the new N-substituent by use of sodium borodeuteride.
N-monoalkylation is not exactly what we want but still, maybe it could give us more clues about the dialkylation and pictet-splenger issue.
http://www.poedecoder.com/Qrisse/works/d
(Stranger)
07-06-03 21:13
No 445093
an other way of decarboxylation of thryptphan is:
tryptophan+cyclohexanol+2-cyclohexen-2-o
see,
A novel decarboxylation af aminoacids,
A facile method of decarboxylation by the use of 2-cyclohexen-2-one as a catalyst,
chemistry letters 1986/893-896
The chemical society of Japan
or you could try:
syntheses of tryptamine HcL from tryptophan.
polish patent PL 130769
yield 80-85 %
(Moderator)
07-07-03 01:51
No 445161
Ann. Chim. = Ann. Chim. (Rome)
(Hive Bee)
07-07-03 03:10
No 445172
an other way of decarboxylation of thryptphan is:
tryptophan+cyclohexanol+2-cyclohexen-2-o
Thanks, but I already know this. This thread's subject is finding a route for N,N-dimethyltryptophan, not about the decarboxylation of tryptophan.
http://www.poedecoder.com/Qrisse/works/d
(Hive Bee)
07-07-03 13:14
No 445300
(Rated as: excellent)
Giancarlo Verardo, Paola Geatti, Elena Pol, and Angelo G. Giumanini
Abstract: alpha-amino acids and alpha-amino methyl esters are easily converted to their N-monoalkyl derivatives by a reductive condensation reaction using several carbonyl compounds in the presence of sodium borohydride. The reaction allows the alpha-monodeuterium labeling of the new N-methyl substituent by use of sodium borodeuteride.
Keywords: alpha-amino acid, alpha-amino methyl esters, sodium borohydride, reductive N-monoalkylation, carbonyl compounds
(The article is 9 pages of text, so I'll only type the interesting parts
Introduction
(First part of the text covers discussion of peptoid synth and old more complicated routes to N-alkyl amino acids/esters.)
Reductive N-alkylation of amino acids and amino esters is a method widely used to synthesize the corresponding N-benzyl derivatives: NaBH(OAc)3 14, NaHTe 15, NaBH3CN 16 and NaBH4 12c, 17 were the reducing agents employed. Sodium triacetoxyborohydride has also been used to reduce imines derived from the reaction between alpha-keto esters and benzylamine 18. The last two reactants showed a larger range of applicability: in fact, they were used with carbonyl compounds (other than benzaldehyde), but sodium cyanoborohydride had to be used under inert atmosphere. Moreover, long reaction times (16-20 h) with amino acids 19 and large excess of carbonyl compounds and the reducing agent with amides of amino acids were needed. Sodium borohydride, on the other hand, has been used in water only on amino acids 12e but, even when the reaction was carried out in the presence of large excess of reactants, yields were not satisfactory.
Results and discussion
On the basis of knowledge we have acquired on the applications of sodium borohydride as reducing agent in the reductive N-alkylation of amines 21, we have now extended its use to alpha-amino acids (1) and alpha-amino methyl esters (2) in combination with several carbonyl compounds (3) (Scheme 1)
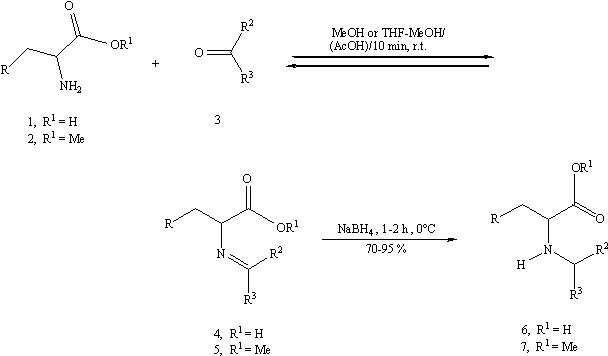
| 1 | R | 2 | R | 3 | R2 | R3 |
| a | H | a | H | a | H | Et |
| b | Me2CH | b | Me2CH | b | H | Pr |
| c | Ph | c | Ph | c | H | tert-Bu |
| d | 4-HO-C6H4 | d | 4-HO-C6H4 | d | H | Ph |
| e | indolyl | e | indolyl | e | Me | Me |
| f | CH2CO2H | f | CH2CO2Me | |||
| h | imidazolyl | |||||
| i | CH2CONH2 | |||||
| j | CH2SMe |
| 6 | R | R2 | R3 | 7 | R | R2 | R3 |
| ab | H | H | Pr | ad | H | H | Ph |
| bb | Me2CH | H | Pr | bb | Me2CH | H | Pr |
| cb | Ph | H | Pr | bd | Me2CH | H | Ph |
| cc | Ph | H | tert-Bu | ca | Ph | H | Et |
| cd | Ph | H | Ph | cb | Ph | H | Pr |
| ce | Ph | Me | Me | cc | Ph | H | tert-Bu |
| dd | 4-HO-C6H4 | H | Ph | cd | Ph | H | Ph |
| eb | Indolyl | H | Pr | ce | Ph | Me | Me |
| ec | Indolyl | H | tert-Bu | db | 4-HO-C6H4 | H | Pr |
| ed | Indolyl | H | Ph | dd | 4-HO-C6H4 | H | Ph |
| fb | CH2CO2H | H | Pr | ea | Indolyl | H | Et |
| fd | CH2CO2H | H | Ph | ec | Indolyl | H | tert-Bu |
| ha | imidazolyl | H | Et | ed | Indolyl | H | Ph |
| hb | imidazolyl | H | Pr | ee | Indolyl | Me | Me |
| hd | imidazolyl | H | Ph | fa | CH2CO2Me | H | Et |
| ib | CH2CONH2 | H | Pr | fc | CH2CO2Me | H | Ph |
| jb | CH2Sme | H | Pr | fd | CH2CO2Me | H | Ph |
| fe | CH2CO2Me | Me | Me |
http://www.poedecoder.com/Qrisse/works/d
(Hive Bee)
07-07-03 13:31
No 445304
(Rated as: good read)
The carbonyl compound (3) was added at room temperature to a methanolic solution of 1 or a hydrochloride of 2 after its neutralisation in situ with a shlight excess of finely powdered NaOH. After 10 min, a time sufficient to form the imine 4 or 5, the reaction mixture was cooled to 0°C and solid NaBH4 was added. GC-MS analysis of the reaction before the addition of NaBH4 indicated that higher temperatures and (or) longer contact times between 2 and 3 did not improve the formation of 5; moreover, the conversion into the corresponding N-alkylated compounds 7 was lower. Since our goal was the use of NaBH4 as the reducing agent in the reductive alkylation of both 1 and 2, the physical form (pellets or granular) of the reductant and minor but essential procedure variations were found to be very important.
The reaction between the amino acids (1) and all carbonyl substrates (3) was carried out in MeOH using NaBH4 pellets (Method A). Unlike water12e, this solvent, was able to solubilize all the species initially present and the species eventually forming in the process, and it also made the NaBH4 reactive enough to reduce the imine 4. The methanolic mixture obtained was acidified with 37% aq HCl up to the isoëlectric point of the starting amino acid (1) and, after the removal of solvent and addition of acetone, a solid was obtained composed of a mixture of the N-alkyl amino acids (6) and NaCl as determined by 1H and 13C NMR and by qualitative analysis. To eliminate NaCl, the solid was washed with water, but unfortunately, all the N-alkyl derivatives of glutamic acid (1f) and, particularly, histidine (1h) were found to be widely soluble in water. For this reason the yield of N-butyl- (6fb), N-benzyl- (6fd) glutamic acid, N-propyl- (6ha), N-butyl- (6hb), and N-benzylhistidine (6hd) were also determined by 1H NMR using phenylacetic acid as internal standard (table 1). This acid, as sodium salt in D2O-NaOH, exhibited a singlet at delta = 3.54, which was far away from all the other signals.
The use of acetone during the work-up of the reaction provided a solid that, freed from all unreacted 3 and (or) the corresponding alcohol, happened to be less soluble in water during the elimination of NaCl.
N-isopropyl phenylalanine (6ce) was obtained only in 63% isolated yield; however, a large excess (3 mol eq) of acetone (3a) was used.
When propanal (3a) or butanal (3b) were used in combination with 2, Method A was followed but granular NaBH4 was added (Method B). These aldehydes reacted very quickly with 2, therefore excess granular NaBH4 was used to promote the reduction of 3a or 3b, which in turn caused the N,N-dialkylation of 2 as shown by the GC-MS analysis of the reaction mixture.
All N-alkyl amino acids were isolated with 63-94% yields.
All N-alkyl-amino acid methyl esters were isolated with 70-95% yields.
When sterically hindered 2,2,2-trimethylacetaldehyde (3c) or benzaldehyde (3d) were used in connection with 2, the addition of acetic acid (1 mol equiv) and the less reactive pelletised NaBH4 were found to be beneficial to the reaction (Method C). In fact, under these experimental conditions, carbonyl compounds (3c) and (3d) and imine 5 are more reactive towards 2 and NaBH4, respectively, because they are partially protonated.
The reaction of 2 with acetone (3e), the least reactive carbonyl compound considered, required more acetic acid (2 mol equiv) than Method C. In addition, THF was added to reduce the reactivity of pelletized NaBH4 towards 3e (Method D).
With the sterically hindered and (or) less electrophilic 2,2,2-trimethylacetaldehyde (3c), benzaldehyde (3d), and acetone (3e), GC-MS analysis sometimes showed the presence of unreacted 2 or 5. A further addition of 3 and NaBH4, or NaBH4 alone (see experimental[i]) was used to bring the reaction to completion. The results of the reactions involving 2 are listed in table 2.
When NaBD4 was used instead of NaBH4, a deuterium atom was introduced in the [i]alpha position of the entering N-substituent. This procedure was succesfully employed in the synthesis of N-(1-deuterio)butyl phenylalanine (6cb-d) and N-(alpha-deuterio)benzyl phenylalanine methyl ester HCl. (7cd-d)
The mass spectra (EI positive ion at 70 eV) of some N-alkyl derivatives of 6 and 7 showed the presence of the [M + 1]+ peak (tables 3 and 4) and the almost complete absence of the parent ion, as invoked to explain the behaviour of substituted malonic acids (22).
Conclusions
In conclusion, the method described here is very simple, fast, and inexpensive, with respect to both the handling of chemicals and the equipment required. It is also applicable both to amino acids and amino acid methyl esters with various carbonyl compounds, and the preformation and isolation of the imine is not required. It appears essential, however, to find the most suitable conditions for succesful experimentations.
Experimental
Method A
N-Alkyl-amino acids (6)
The aldehyde (3a-d, 14.0 mmol) was aded to a stirred solution of the amino acid (1, 10.0 mmol) in MeOH (25 mL) neutralised with finely powdered NaOH (0.42 gr, 10.5 mmol). After 10 min at room temperature, the solution was cooled with an ice bath and NaBH4 pellets (0.49 gr, 13.0 mmol) were added. The reaction mixture was stirred for 120 min at 0°C, then acidified with 37% HCl up to the isoëlectric point of the starting amino acid 1. The solvents were distilled off and the residue was triturated in acetone and filtered. The solid was washed with H2O to remove NaCl and pure N-alkyl amino acid 6 was obtained.
With tyrosine (1d) and glutamic acid (1f), twice the amount of NaOH (0.84 gr, 20.1 mmol) was used to neutralise all the acid centers.
When acetone 3e wes used as the carbonyl compound, the procedure was the same, but 30.0 mmol (2.20 mL) of 3e was used.
Method B
N-propyl and N-butyl amino acid methyl ester hydrochlorides (7)
Propanal or butanal (3a or 3b, respectively, 14.0 mmol) was added to a stirred solution of the amino acid methyl ester HCl. (2, 10.0 mmol) in MeOH (50 mL) neutralized with finely powdered NaOH (0.42 gr, 10.5 mmol). After 10 min at RT, the solution was cooled with an ice-bath and granular NaBH4 (0.30 gr, 8.0 mmol) was added. The reaction mixture was stirred at 0°C for 60 min, then MeOH was distilled off, the residue re-dissolved in EtOAc, and NaCl filtered off. The organic phase was washed with brine and dried over Na2SO4. EtOAc was removed in vacuo and the residue, dissolved in Et2O, was treated with ethereal HCl (2M, 5 mL, 10mmol); N-alkyl amino acid methyl ester HCl. 7 was filtered and washed with Et2O. If the residue was insoluble in Et2O, it was dissolved in EtOAc, treated with ethereal HCl and, after elimination of solvents under reduced pressure, 7 was crystallised from Et2O.
Method C
N-benzyl and N-neopentyl amino acid methyl ester hydrochlorides (7)
2,2,2-trimethylacetaldehyde or benzaldehyde (3c or 3d, respectively, 15.0 mmol) and acetic acid (0.57 mL, 10.0 mmol) were added to a stirred solution of the amino acid methyl ester HCl. (2, 10 mmol) in MeOH (50 mL) neutralized with finely powdered NaOH (0.42 gr, 10.5 mmol). After 10 min at RT, the solution was cooled with an ice-bath and NaBH4 pellets (0.38 gr, 10.0 mmol) were added. The reaction mixture was stirred at 0°C for 60 min, and if the GC-MS analysis of the reaction mixture showed the presence of unreacted imine 5, granular NaBH4 (0.19 gr, 5.0 mmol) was added. The reaction mixture was stirred for an additional 30 min, then MeOH was distilled off, the residue dissolved in EtOAc, and NaCl filtered off. The organic phase was washed with saturated Na2CO3 solution and dried over Na2SO4. EtOAc was removed in vacuo and the residue, dissolved in ether, was treated with ethereal HCl (2M, 5 mL, 10.0 mmol); N-alkyl amino acid methyl ester HCl. 7 was filtered and washed with ether. If the residue was insoluble in ether, it was dissolved in EtOAc, treated with ethereal HCl and, after the elimination of solvents under reduced pressure, 7 was crystallised from ether.
Method D
N-isopropyl amino acid methyl ester hydrochloride
A solution of acetone (3e, 1.47 mL, 20.0 mmol) and acetic acid (1.14 mL, 20.0 mmol) in THF (6 mL) was added to a stirred solution of amino acid methyl ester HCl. 2 (10.0 mmol) in MeOH (18 mL) and THF (35 mL) neutralised with NaOH (0.42 gr, 10.5 mmol). After 10 min at RT, the solution was cooled with an ice-bath and NaBH4 pellets (0.38 gr, 10.5 mmol) were added. The reaction mixture was stirred for 60 min at 0°C and if the GC-MS analysis of the reaction mixture showed the presence of the unreacted amino acid ester 2, a solution of acetone (3e, 0.73 mL, 10.0 mmol) and acetic acid (0.57 mL, 5.0 mmol) in THF (3 mL), and after 10 min NaBH4 pellets (0.19 gr, 5 mmol), were added. The reaction mixture was stirred for an additional 30 min, then the solvents were distilled off, the residue was dissolved in EtOAc, and NaCl was filtered off. The organic phase was washed with saturated Na2CO3 solution and dried over Na2SO4. EtOAc was removed in vacuo and the residue, dissolved in ether, was treated with ethereal HCl ( 2 M, 5 mL, 10.0 mmol); N-isopropyl amino acid methyl ester HCl. 7 was filtered and washed with ether. If the residue was insoluble in Et2O, it was dissolved in EtOAc, treated with ethereal HCl and, after the elimination of solvents under reduced pressure, 7 was crystallized from Et2O.
References
12c : P. Quitt, J. Hellerbag and K. Vogler, Helv. Chim. Acta 46, 327 (1963)
12e : E. Davidov, H. Spiegelstein-Klarfeld, O. Gisri-Yaron, E. Naim and N. Lichtenstein, [blue]Isr. J. Chem., 7, 487 (1969
14a : B.A. Kulkarni and A. Ganesan, Angew. Chim. Int. Ed. Engl. 36, 2454 (1997)
14b : D.L. Boger, J. Zhou, R.M. Borzilleri, S. Nukui and S.L. Castle, J. Org. Chem. 62, 2054 (1997)
14c : B. Cao, D. Xiao and M.M. Jouillie, Org. Lett. 1, 1799 (1999)
15a : D. Zhou, Y. Guan and S. Jin, Chem. Lett. 1, 209 (1990)
15b : D. Zhou, Y. Guan and S. Jin, Bejing Daxue Xuebao Ziran Kexueban 29, 428 (1993)
16a : J. Bastide, C. Coste and J-L Marty, C.R. Hebd. Seances Acad. Sci. Ser. C, 287, 471 (1978)
16b : S. Laschat, R. Frohlich and B. Wibbeling, J. Org. Chem. 61, 2829 (1996)
16c : A. Nefszi, J.M. Ostresh and R.A. HOughten, Tet. Lett. 38, 4943, (1997)
17a : D. Soerens et al, J. Org. Chem. 44, 535 (1979)
17b : T.J. Cheeseright et al, J. Chem. Soc. Perkin Trans. I, 1595 (1994)
17c : M. Weighl and B. Wunsch, Org. Lett. 2, 1177 (2000)
18 : A. Abdel-Magid et al, J. Org. Chem. 61, 3849 (1996)
19 : Y. Ohfune et al, Chem. Lett. 441 (1984)
21a : A.G. Giumanini et al, J. Labelled Compd. Radiopharm. 24, 255 (1987)
21b : G. Verardo et al, Synthesis, 447 (1991)
21c : G. Verardo et al, Synthesis, 121 (1993)
21d : G. Verardo et al, Synth. Commun. 24, 609, (1994)
21e : G. Verardo et al, Synthesis, 74 (1999)
http://www.poedecoder.com/Qrisse/works/d
(Moderator)
07-07-03 14:12
No 445318
Regarding the tables: there has to be the same number of cells (td) in every row (tr).
Are there any hard facts in the article that they didn't cyclize Trp and got good yields?
(Hive Bee)
07-07-03 14:44
No 445329
Are there any hard facts in the article that they didn't cyclize Trp and got good yields?
The lowest yield they've got was 63%, for N-isopropyl phenylalanine. Apparantly acetone is the least reactive carbonyl compound in their experiments. The other reactions that used aldehydes as carbonyl compounds always yielded between 73-94% for the amino acids.
They've got N-butyl, N-neopentyl and N-benzyl tryptophan in 82%, 90% and 94% yield respectively.
Of course that still says nothing about using formaldehyde.
http://www.poedecoder.com/Qrisse/works/d
(Hive Bee)
07-07-03 21:41
No 445470
I've got two thoughts on that. One, it gets turned into DMT or alternatively, bufotenine.
has anyone bioassayed this novel amino acid?
you can't keep peace with a gun
(Hive Bee)
07-08-03 01:22
No 445524
Ingestion of N,N-dimethyltryptophan, although it's pharmacological action is unknown, will most probably not be psychoactive.
Here is what Dr. Shulgin says: (http://www.cognitiveliberty.org/shulgin
In principle, the effectiveness of the body's amine group remover, the monoamineoxidase enzyme, is blocked by a lumpy group (such as a methyl group) located right next to it. But this is not a methyl group; it is a monster negatively charged carboxy group which effectively destroys the innocent amine character of that tryptamine nitrogen atom. The compound you mention would be a full fledged amino acid and, as such, would be blocked from entry into the brain by its hydrophilic salt nature.
The best way to administer this compound would be by smoking/vaporising it, as there is a good chance than N,N-dimethyltryptophan will decarboxylate when heated above its melting point.
http://www.poedecoder.com/Qrisse/works/d
(Hive Bee)
07-08-03 01:33
No 445526
I posted up the results of an experiment I did many moons ago with lysine and tyrosine using a sealed pipe with acetone and the amino in an oven at 250°C, and the results certainly seemed to be a significant proportion of transformation into freebases, but nobody's verified it (*aghast* and with all their chemistry equipment too).
can nn dimethyl T be bought from chem suppliers? wait a mo... nah, can't find it at the tryptamine specialists.
heh, but if anyone finds a supplier of nn dm T, damn, pm me, cos that would be way cool
you can't keep peace with a gun
(Hive Bee)
07-08-03 18:00
No 445723
When propanal (3a) or butanal (3b) were used in combination with 2, Method A was followed but granular NaBH4 was added (Method B). These aldehydes reacted very quickly with 2, therefore excess granular NaBH4 was used to promote the reduction of 3a or 3b, which in turn caused the N,N-dialkylation of 2 as shown by the GC-MS analysis of the reaction mixture.
What the researchers say here, is that when they employed Method A using propanal or butanal on a amino acid methyl ester, these aldehydes reacted very quickly with the AA methyl ester, and excess granular NaBH4 used to promote the reduction of these aldehydes caused N,N-dialkylation of the amino acid methyl ester.
We see that Method A uses 14.0 mmol of aldehyde, 14.0 mmol of AA methyl ester and 13.0 mmol of NaBH4 (pellets), and the mixture was stirred for 120 min.
Method B, however, employed when propanal or butanal were used as the carbonyl compounds, uses only 8.0 mmol of NaBH4, and they let the mixture stir for only half the time, to get good yield of N-alkylated product.
Note that propanal and butanal are the lowest chained carbonyl compounds utilized in this paper, formaldehyde nor acetaldehyde were never tried here.
Could it be that N-alkylation is only achieved when the carbonyl compound is large or sterically hindered enough as to make N,N-dialkylation difficult or impossible? Thus that formaldehyde and acetaldehyde were never used because they would always result in N,N-dialkylation?
So, would it be a good idea to try Method A with tryptophan and formaldehyde, perhaps with an excess of aldehyde and NaBH4, so that N,N-dialkylation would be the main reaction?
Another question about the Pictet-Spengler reaction: doesn't this only occur in acidic medium? Here the amino acid (methyl ester) is always neutralized with NaOH, is it still possible for it to cyclisize then? It is nowhere mentioned in the paper.
Hey, maybe I should e-mail the author of the paper some questions? ![]()
A Dream Within A Dream (http://www.poedecoder.com/Qrisse/works/
(Hive Bee)
07-10-03 07:22
No 446193
(Rated as: excellent)
The chances that tryptophan will cyclisize due to the Pictet-Spengler reaction in the HCHO/NaBH4 alkylation are IMHO very low, considering the low temperature the reaction is kept at and the small amount of time that is necessary to complete it.
"Pictet-Spengler reactions in aprotic media", Mikolaj Jawdosiuk and James W Cook, J. Org. Chem. (1984), 49, 2699-2701 ,
In 1976 we reported that reaction of tryptophan methyl ester (1) with aldehydes such as benzaldehyde (2a), cyclohexanecarboxaldehyde (2b), or alpha-keto acids in refluxing benzene (Dean-Stark trap to remove water)1 provided much improved yields of the Pictet-Spengler reaction with respect to the traditional reaction performed in aqueous, acidic media. The reasons for this were very simple for acid-labile substrates were much less prone to decomposition in a non-acidic, nonaqueous medium.
Since our original reports, 1,2 a number of 3-methoxycarbonyl tetrahydro-beta-carbolines have been succesfully prepared by this procedure 3,5-7,11. In view of these reports it was suprising to find that Grigg et al. reported that "A repeat of Cook's original work (tryptophan methyl ester, benzaldehyde, benzene, 80°C, 48h), i.e., generating the Schiff's base in situ gave only Schiff's base (1a) and no beta-carboline (2a,b)"4.To examine the conflicting experiences regarding this reaction, we have carried out several further experiments.
An important feature of the procedure that was succesful in our hands is use of a Dean-Stark trap below the reflux condensor to remove water from the reaction1,2.
In the Grigg report, most of the experiments were carried out in sealed NMR reaction tubes, and no mention is made of the use of a water separator.4 We have compared the course of the reactions of 1 and benzaldehyde (2a, purified by K2CO3 wash, drying, distillation) in benzene with an open system and a water separator, and in refluxing benzene in a closed system. Under the former conditions, after 12 h TLC indicated the presence of about 50% imine 4a, the remainder of the material was a mixture of cis and trans carbolines 5a. After 48 h the reaction had proceeded almost completely to 5a. In a closed system without removal of water, the formation of 5a was negligible, and the Schiff's base 4a was recovered quantitatively (Scheme 1).
Scheme 1:

To definitely determine the significance of the use of a Dean-Stark trap in the sequence, identical reactions btween 1 and 2a were performed both open to the air; however, in one case a Dean-Stark trap was used, while in the second experiment none was employed. After 24 h at reflux, aliquots of each reaction were examined by 13C NMR. The condensation performed in the usual manner (DST) gave almost exclusively tetrahydro-beta-carboline 5a at this point, whereas the reaction carried out in the absence of the trap gave exclusively the non-cyclisized 4a. Nevertheless cyclization in the absence of a DST did eventually take place for after 84 h of heating this gave the cyclized product 5a, but the reaction was extremely slow in comparision to the previously reported conditions (DST).1,2
A second point concerning reaction conditions concerns the presence of acid. Grigg et al. demonstrated the role of acid catalysis in the reaction and suggests that acidic impurities are responsible for succesful cyclization. To test the possibility that air oxidation of aldehyde occurred under the conditions originally used, the reaction of 1 and 2a was carried out in a nitrogen stream, and cyclization to 5a was found to be much slower. Moreover, addition of the proton-scavenger DBU9 completely suppressed the cyclization. The weaker base imidazole retarded the reaction significantly. Similar results were observed in reactions of 1 with the more reactive cyclohexanecarboxaldehyde (2b). These data support the hypothesis that a small amount of benzoic acid formed by air oxidation facilitates the cyclization.
In regard to the report by Grigg et al.4 that the ratio of cis and trans isomzers of the 1-phenyl-3-(methoxycarbonyl)-1,2,3,4-tet
In conclusion, we emphasise that our experience and that of several other groups3,5-7,11 establishes, contrary to the implication of Grigg et al,4 that the simple condensation of tryptophan methyl ester and aldehydes in refluxing benzene in an open system with a water separator provides a simple and very practical method for preparing 3-(methoxycarbonyl)-1,2,3,4-tetrahydro-beta-carbolines.
Tryptophan methyl ester was obtained from dl-tryptophan by esterification in methanolic HCl and converted into the freebase (K2CO3).12 This material 1 was recrystallised from ether-hexane (mp. 70.5-72°C (lit. mp.13 71-73°C)). NMR spectra were recorded on Varian T-60 MHz, Varian CFT-20 13C NMR and Bruckner WH-250 (250 MHz Fourier transform instrument with multinuclear capability) spectrometers. Precoated TLC sheets used were silica gel 60F-254 (E. Merck). TLC plates were developed with the spray reagent ceric ammonium sulfate in 50% sulfuric acid.
Reactions of tryptophan methyl ester (1) with benzaldehyde (2a). Preparation of Nb-Benzylidenetryptophan methyl ester (4a).
Tryptophan methyl ester (1, 4.4 gr, 0.02 mol) and benzaldehyde (2a, 2.5 gr, 0.025 mol) were dissolved in benzene (70 mL) and the solution was refluxed for 1 h in a flask equipped with a Dean-Stark trap and a reflux condensor open to air. The solvent was evaporated and the residue was recrystallised from metrhanol to afford 5.87 gr material 4a which melted at 123-126°C (95.7% yield). An analytical sample melted at 130°C (lit. mp. 120°C1, 128-129°C4); 13C NMR (CDCl3, Me4Si) 29.7, 52.1, 73.8, 111.2, 118.8, 119.4, 122.0, 123.6, 127.4, 128.5, 131.0, 135.8, 136.3, 163.3, 172.7.
Direct preparation of 3-(methoxycarbonyl)-1-phenyl-1,2,3,4-tet
1. Tryptophan methyl ester (1, 2.2 gr, 0.01 mol) and benzaldehyde (2a, 1.1 gr, 0.0105 mol) from a newly purchased bottle were dissolved in distilled benzene (50 mL). The solution was held at reflux under a reflux condensor (open to air) equipped with a Dean-Stark trap. The reaction progress was monitored by TLC after 12, 24, 48 and 70 h by comparision of Rf values with those of authentic samples2. After 12 h about 50% of the Schiff base was converted into a cis/trans mixture of 5a. After 24 h the conversion increased to about 70-85% (estimated by TLC). After 48 hours the reaction mixture showed only a small amount of unreacted 4a (TLC) and after 70 h the reaction was essentially completed. Evaporation of benzene to dryness gave 3.19 g of crude 5a. TLC of this material showed only two spots corresponding to the cis and trans isomers of 5a. The chemical shifts of the carbon atoms in the 13C NMR spectrum of the material were consistent with those reported in the literature8. The crude product was dissolved in CDCl3 and the 1H and 13C NMR spectra were taken. The integration of the C(1) protons in the cis and trans 5a isomers (250 MHz NMR spectrometer) was consistent with a ratio of cis to trans of 58:42 while integration of the 13C NMR spectrum with suppressed NOE for several superimposable resonance lines also gave the value of 58:42. These values are consistent with values reported in the literature (54:46) measured with mass spectroscopy3 and (54.5:45.5)4 measured by 90 MHz 1H NMR.
2. The reaction described above (see 1) was carried out in an open system exactly analogous to the above experiment but in absence of a Dean-Stark trap (water separator). After 24 h an aliquot of the reaction mixture was analysed by 13C NMR and was shown to essentially contain only the imine 4a. On heating the mixture for 36 h the tetrahydro-beta-carbolines began to appear (TLC +- 30% 5a) and after 84 h 4a had cyclized completely to 5a. The control experiment (experiment 1, DST) after 24 h at reflux contained essentially only 5a (13C NMR analysis).
3. The same quantities of 1 and 2a and benzene were used as in the two previous cases, however, 2a was washed with a 50% solution of K2CO3 and then dried (MgSO4) and distilled under reduced pressure at 38°C (1 torr). The reaction mixture was held at reflux for 84 h in a closed system with a reflux condensor in the absence of a Dean-Stark trap. The condensor was closed with an oil bubbler. After 84 h TLC did not show any appreciable amounts of the cyclized product 5a. Evaporation of benzene to dryness gave a qsuantitative recovery of 4a.
4. The reaction sequence termed 3 above was repeated by employing a Dean-Stark trap but under a nitrogen (Ameri-Gas 99%) atmosphere. After the reaction was held for 12 h in refluxing benzene, no trace of 5a was observed (compare 50% in experiment 1), and after 48 h only 10% of 5a had been formed.
5. The reaction sequence termed 1 as above was carried out in the presence of 1.5 g (0.01 mol) of DBU (1,8-dizazbicyclo-[5.4.0]undec-5-ene) although 2a was pretreated as indicated under 3. After 70 h no 5a was detected. Although DBU is a strong enough base9 to act as a proton scavenger and also abstract proton from the alpha position of the ester, the Schiff base was observed in this sequence by TLC.
The same reaction was repeated (reagents the same as above) with addition of 0.7 g (0.01 mol) of imidazole. After the mixture was held for 50 h in refluxing benzene, the amount of cyclized product 5a was estimated to be 10-15%
References
1: Sandrin J et al, Heterocycles 4 (1976), 1101
2: Soerens, D et al, J. Org. Chem. (1979) 44, 535
3: Kumar, S; Seth, M; Bhaduri A P, Ind. J. Chem. B (1981) 1078
4: Grigg R et al, J. Chem. Soc. Perkin Trans. I (1983) 185
5: Harisson, D M ,Tetrahedron Lett. 22, (1981) 2501
6: Shimizu, M et al, Chem. Pharm. Bull. 30 (1982) 3435
7: Toyoda, Y et al, Chem. Lett. (1982) 903
8: Ungemach, F et al, J. Am. Chem. Soc. 102 (1980) 6976
9: DBU has been employed to deprotonate malonic esters for alkylation reactions and therefore should serve as a good proton scavenger in this case (Oedigerod, H and Moeller, F, Liebigs Ann. Chim. (1976) 348). It is also a strong enough base to deprotonate trinitrotoluene (Sugimoto, N. et al, J. Phys. Chem. 86 (1982) 3418; Ebel, H, F. C-H Acidity of organic compounds in "Methods der Organischen Chemie, Houben-Weyl, 1970, 13 (part I), 27, 57).
10: Hamaguchi, F; Nagasaka, T; Ohki, S, Yakugaku Zasshi (1974) 94, 351
11: Kumar, S et al, Ind. J. Chem. (1983) 22B, 54
12: Otsuka, H & Inouye, K, Bull. Chem. Soc. Jpn. 37 (1964) 1465
13: Brenner, M; Sailer, E; Kocher, V, Helv. Chim. Acta 31 (1948) 1908
A Dream Within A Dream (http://www.poedecoder.com/Qrisse/works/
(Moderator)
07-10-03 08:29
No 446208
I have a cool idea: quaternary trimethyl-alkyl-amines are converted into dimethyl-alkyl-amines with LiAlH4. Maybe that's also possible with NaBH4!
Then you could take tryptamine or tryptophan, let it stand with methyl iodide for a while, and let the product react with NaBH4 to get the dimethylamino analog (the resulting tryptophan-methyl-ester would have to be hydrolyzed).
(Hive Bee)
07-10-03 10:25
No 446234
I've always wondered why dr. Shulgin used LiEt3BH.
Perhaps because THF is used as the solvent, NaBH4 is practically insoluble in THF (1gr/L).
Maybe we could just reflux the quaternary salt with NaBH4 in MeOH or EtOH?
A Dream Within A Dream (http://www.poedecoder.com/Qrisse/works/
(pHantasticant)
07-11-03 05:35
No 446410
Thank you for those refs Post 445304 (Vitus_Verdegast: "continued.", Tryptamine Chemistry)!
If your dream come true, you must say: http://www.dailywav.com/1000/children.wa
Accept No Imitations, There Can Only Bee One; www.the-hive.ws
(Chief Bee)
07-11-03 13:31
No 446451
If you add one equivalent of LiCl to a suspension of NaBH4 in THF, the soluble LiBH4 is formed in situ, and as it is a stronger reedicing agent than NaBH4, the reduction is even more likely to happen.
(Moderator)
07-11-03 16:11
No 446486
BTW, to get a good yield of the quaternary amine you have to add an equimolar amount of a more-basic, non-nucleophilic base (like di-isopropyl-ethyl-amine) as an acid scavenger.
(Hive Bee)
07-13-03 07:36
No 446814
Found some disencouraging info on the pyrolysis of tryptophan :
http://www.healthplanning.gov.bc.ca/guil
(BATCo document for Province of British Columbia, 8 November 1999)
Chemical Identity of Some of the Mutagenic Products of Protein Pyrolysis
The majority of work has centered on identifying the mutagens from the pyrolysed amino acid tryptophan, which represent some of the most powerful mutagens known to date (3, 4). From tryptophan pyrolysate, the mutagens 3-amino-1,4-dimethyl-5H-pyrido(4,3-b)ind
ole and 3-amino-l-methyl-5 H-pyrido(4,3-b)indole have been isolated (4). These compounds are both gamma-carbolines (Figure 1) and have also been found in pyrolysis products of whole proteins (7).
Furthermore, it is well known that the co-mutagens harman and norharman (beta-carbolines) from tryptophan pyrolysis are also present in the [cigarette smoke] condensate (Figure 1). All these compounds have complex mutagenic activities. The amino-alpha-carbolines are themselves powerful mutagens and also act synergistically with other mutagens (9) whilst the beta-carbolines are well established co-mutagens*(4).
Figure 1
*Co-mutagens are not themselves mutagenic, but are able to enhance the mutagenicity of other compounds. (BATCo document for Province of British Columbia 8 November 1999)
3. Matsumoto, T., Yoshida, D., Mizusaki, S. and Okamoto, H. Mutagenic activity of amino acid pyrolysates in Salmonella typhimurium. TA98. Mut. Res. 48, 279-286, (1977).
4. Sugimura, T. and Naigo, M. Mutagenic factors in cooked foods. CRC Critical Reviews in Toxicology 6, 189-209, (1979).
7. Yoshida, D., Matsumoto, T and Nishigata, H. Effect of heating methods on mutagenic activity and yield of mutagenic compounds in pyrolysis products of protein. Agric. Biol. Chem. 44, 253-255, (1980).
9. Yoshida, D. and Matsum to, T. Amino-a-carbolines as mutagenic agents in cigarette smoke condensate. Cancer Letters 10, 141-149, (1980)


http://www.hplc1.com/shodex/english/dc01
Medline (PMID=9641497) says:
Cooking meat creates heterocyclic amines (HCAs) through pyrolysis of amino acids and creatinine.
The question is: what will happen if pure tryptophan or N,N-dimethyltryptophan are subject to pyrolysis?
A Dream Within A Dream (http://www.poedecoder.com/Qrisse/works/
(Hive Bee)
07-23-03 13:47
No 449324
(Rated as: good read)
Well, I heard that Hjalmar P., a Hungarian architect, performed this reaction a while ago
To a stirring solution of L-tryptophan (2 g, 10 mmol) and NaOH (0.42 g, 10.5 mmol) in 25 mL MeOH was added a 30% formaline solution (3 g, ~30 mmol). After 15 min of stirring at RT (~25°C), the mixture was cooled in an ice/salt bath and NaBH4 (600 mg, 15 mmol) was added bit by bit, care was taken that the temperature did not rise above 5°C. After an hour, all ice was melted, and the solution was stirred overnight.
It was then carefully acidified with HCl, to the iso-electric point of tryptophan (pH 5.89), where a small amount of solid, unreacted L-tryptophan, precipitated. This was filtered off, and the MeOH was evaporated in vacuo. An attempt was done to wash the remaining solid with acetone, but after the acetone was evaporated it left nothing.
The precipitate was extracted with a small amount of hot ethanol, (what was presumed to be) NaCl remained undissolved and was filtered off, and the warm EtOH was allowed to come to RT. After sitting overnight in the freezer, a offwhite precipitate formed.
This was filtered and the precipitate was air-dried, it weighed 0.5 g. Evaporation of the alcohol left 0.7 g of yellow substance which was gummy at first, but quickly became a hard solid.
The Harvey, Miller and Robson test was employed according to J. Chem. Soc. (1940) p. 154-155:
A small amount of the substance was dropped in a test-tube containing sulphuric acid and a trace of oxidizing agent. The paper suggests to use FeCl3 among others, but in this case 50% H2O2 was used. Tryptophan, its methyl ester and N-methyltryptophan gave a colour reaction within a couple of minutes from pale pink -> weak purple -> yellow with strong fluorescence, so does tryptamine.HCl but with a less marked fluorescence. Most beta-carboline-4-carboxylic acids give a blue colour
Both obtained substances were tested this way, together with pure L-tryptophan. All three gave the same colour reactions, ascribed to tryptophan.
The melting point still has to be obtained. These melting points were found in the literature: by Chimimanie, thanks
L-tryptophan : 289°C
N-Methyltryptophan : mp ranges over 100° (!)
N,N-dimethyltryptophan : 239-243°C
2,3,4,5-tetrahydro-beta-carboline-4-carboxylic acid : 306°C
3-methyl-2,3,4,5-tetrahydro-beta-carboline-4-carboxylic acid : 208°C, after softening at 194°C
Also, most beta-carboline-4-carboxylic acids have a tendency to separate after recrystallisation as typical rod-like crystals, which makes identification easier.
Now after reading the above ref (digged up by Chimimanie, thanks again
I.
p. 157
alpha-hydroxymethylamino-beta-3-indolylpropionic acid
l-Tryptophan (0.6 g) was dissolved in water (12 mL), formalin (2 mL) added and the mixture was incubated at 38°C for not more than 3-4 hours. The colourless, crystalline, but rather granular solid which had then separated was collected, washed with cold water, and dried in a vacuum.
Yield 60%. mp 226-240°C
If the above mixture is incubated at 38° for 15 hours in presence of 10 mL 0.1 N NaOH, the beta-carboline is obtained in 80% yield. The beta-carboline is also formed when l-tryptophan is boiled in aqueous solution for 2 hours.
Also, the beta-carboline crystallises from an equimolar aqueous solution of tryptophan and acetaldehyde when kept overnight at RT, according to Harvey and Robson, J. Chem. Soc. (1938) 97.
II.
Reduction of this with NaBH4 as described in the above post yields N-methyltryptophan.
III.
p.158
3-methyl-2,3,4,5-tetrahydro-beta-carboline-4-carboxylic acid
To a cool solution of r-alpha-methylamino-beta-3-indolylpropionic acid (0.5 g) in water (75 mL), a slight excess (0.4 mL; 2.3 mols.) of commercial formalin was added, the whole incubated at 38° for 15 hours. The "solid" which separated was collected (0.11 g), and the filtrate concentrated to a small volume on a boiling water bath. The bundles of stout rods which settled on cooling were collected, washed with absolute alcohol, and dried in a dessicator. Concentration of the mother-liquor gave another crop of the carboline-4-carboxylic acid.
Yield, 0.40 g (76%)
Now, there is a good chance that, if N-methyltryptophan and formaline are incubated for only 2-3 hours at 38° N-hydroxymethyl-N-methyltryptophan will be the main product.
If so, then this can be isolated and again reduced as described before.
A Dream Within A Dream (http://www.poedecoder.com/Qrisse/works/
(Hive Bee)
07-23-03 14:43
No 449332
(Rated as: good read)
From: "beta-2,4,5-Trimethoxyphenylethylamine, an isomer of mescaline", by Max. P. J. M. Jansen.
Recueil des Travaux Chimiques des Pays-Bas et de la Belgique 50, 291-312 (1931). (Edit: Ref corrected by Chimimanie)
p 312:
D. beta-2,4,5-trimethoxyphenylethylamine from 2,4,5-trimethoxyphenylalanine
3 g 2,4,5-trimethoxyphenylalanine were heated rapidly in a small retort over a free flame. The solid sintered together, frothed up and the liquid, which was turbid at first, distilled over along with nauseous smelling fumes
The distillate and the contents of the flask were extracted with dilute HCl, the acid extract made strongly alkaline, extracted with ether, the ethereal solution dried with Na2SO4 and evaporated: yield 0.7 g.
Schaaf and Labouchere have obtained mescaline from 3,4,5-trimethoxyphenylalanine this way (Helv. Chim. Acta 7 (1924) 357)
So, if this is applicable on substituted phenethylamines, I don't see why it shouldn't work for N,N-dimethyltryptophan, but they only got an ~28% yield
I sure hope, in case of DMTrp, these won't be powerful mutagenic pyrazines like the ones obtained from tryptophan pyrolysis, described earlier in this thread.
If I decide to 'pyrolyse' the DMTrp, y'all will be saying : http://www.geocities.com/eric_vornoff/ma
A Dream Within A Dream (http://www.poedecoder.com/Qrisse/works/
(Moderator)
07-24-03 00:05
No 449444
The easiest test would be Ehrlich's reagent, which forms red to blue colors by reaction with the indole-2 (or indole-3 if free) position. In beta-carbolines the 2-position is blocked.
(Hive Bee)
07-27-03 16:52
No 450261
(Rated as: good read)
I asked the author's opinion on this subject,
dr. G. Verardo, personal communication :
Your thoughts are correct, in fact granular NaBH4 is used in Method B because it is more reactive than pellets one so it is able to reduce faster excess aldehyde than the imine. In this way when once the N-monoalkylated compound was formed there was no further carbonyl compound present. With our methods we are not able to obtain the monoalkylation using formaldehyde and acetaldehyde.
Not only is this good news for the subject of this thread, there also could be hope for a tryptamine->DMT using HCHO/NaBH4 synthesis as described in Post 431982 (Lego: "Article on DMT derivate synths", Tryptamine Chemistry).
As I understand it normally NaBH4 is undesirable because it tends to reduce the aldehyde faster than the imine. Now we know that using NaBH4 pellets gives the carbonyl compound ample time to form the imine, so higher yields should be obtained.![]()
A Dream Within A Dream (http://www.poedecoder.com/Qrisse/works/