05-22-03 21:50
No 434889
In Rylander's book ( thx orgk!) he describes a paper which discuss the variations in degree of activity of preparations of Pd/C catalyst's all using the same base materials.
If anyone has library access, the following journal article is the reference for this statement.
J. Org. Chem. 18, 227 (1953) j.g young, w.h. hartung and H.H. Daniels
This should be a very interesting read if someone would be kind enough to post it..
Infinite Radiant Light - THKRA
(Chief Bee)
05-22-03 22:20
No 434895
Palladium Catalysis VI - The Reproducibility of Palladium-on-Charcoal Catalysts (../rhodium/pdf/reproducibility.of.pd-c.catalysts.pdf)
(Hive Bee)
05-23-03 03:53
No 434941
(Rated as: excellent)
A special preparation for a one step (low pressure) reduction of 1-phenyl-2-nitropropene LT/:Edit: to amphetamine (1-phenyl-2-aminopropane)End Edit:
Patent US3458576 [pdf]
(Hive Addict)
05-23-03 05:03
No 434949
Roger I could kiss you!! Thank you for finding that patent
Freaky
(Hive Bee)
06-12-03 07:11
No 439524
(Rated as: good idea!)
And a reactivation method for Pd/C
Patent US3214385: [pdf]
(Daddy)
06-12-03 08:36
No 439531
Basicly: Treat spent catalyst (5%Pd/C) with dilute (5-10%) NaOH or KOH aqueous solution, then washing the catalyst with water, drying the catalyst, and expose the catalyst to air.
Have seen this mentioned several times before here, f.ex. by PolytheneSam?
Still good idea. LT/
WISDOMwillWIN
(Daddy)
06-12-03 09:18
No 439534
that pdf file which Rhodium digged up so fast, it has something to remind however:
""Aside from theoretical considerations, the results of this study serve to em-
phasize the importance of accuracy and precision in catalytic hydrogenation ex-
periments, and indeed, add impetus to the statement of Homer Adkins (3):
�It may not be amiss, however, to suggest that the author of a paper upon
catalytic hydrogenation should state exactly how he prepared his catalyst ; how
much of it and the hydrogen acceptor be used; a t what pressure of hydrogen, at
what temperature, and for how long the reaction was carried out; and how much
of each product was actually isolated in a pure state.� ""
The last link from roger is a 20 years younger one, and there are many recent studies written on this same subject, see the Catalysts Degussa site, http://www.sivento.de/catalysts/ or the Johnson Matthey Catalysts website, or mine around at Espaceweb. LT/
WISDOMwillWIN
(Hive Addict)
06-12-03 09:30
No 439536
Perhaps simple treatment with 2 or 3N sulfuric acid with slight heating (50-60°C) will reactivate Pd or Pt catalyst after amine production too? I've heard that this will be sufficient unless the crystal structure has been changed due to too high temperatures during reaction. Then metal recovery by aqua regia treatment must be done and the catalyst prepared again.
Freaky
(Hive Addict)
06-12-03 11:07
No 439554
(Rated as: excellent)
US Patent 3214385
Reactivation of Spent Pd/C Hydrogenation Catalyst
Abstract:
Catalyst having been used in the manner of this patent can be regenerated using washes with dilute alkali metal hydroxide solution, washing the catalyst with water until the washing have pH of 7.5-10.0, drying the catalyst, and then exposing the catalyst to an oxygen containing gas. This procedure increases the number of times the catalyst may be used without substantial reduction in catalytic activity.
Example 1:
A: (Initial Reaction)
100 parts succinic anhydride, 450 parts ethyl acetate and 8 parts of 4.5% Pd/C were charged to a stainless steel, rocking autoclave. The temperature was brought to 40*C and the agitation started. Hydrogen was introduced to the autoclave to a pressure of 1200 psig. The hydrogenation reacted for one hour and the temperature was allowed to rise to 75*C. The reaction mix was then cooled, filtered through a medium-porosity sintered glass filter to remove the catalyst and then distilled at atmospheric pressure to remove the ethyl acetate and water. The residue was vacuum distilled at to give 79 parts of a fraction boiling at 91-92*C/17mmHg. This corresponds to a 90% of butyrolactone.
B: (Recycling the Catalyst)
The catalyst on the filter was washed with 40 parts of ethyl acetate at RT to remove crude product. After drying for 10 minutes with the aid of suction, the catalyst was slurried with 2x40 part portions of 5% aq. NaOH solution at RT, being filtered after each portion. The catalyst was then slurried for 4 minutes with 2x40 parts of water, between each of which the catalyst was filtered off. The wet catalyst was then dried with filtered air for 10minutes during which time heat was evolved. The catalyst thus obtained was recycled to the autoclave and the hydrogenation above was carried out with the recycled catalyst.
Notes:
The catalyst gave yields of 84-91% of theory in all reactions. (The 84% was achieved after 18 uses of the same catalyst having been run successive reaction/cleaning cycles.) In some cases alcoholic (ethanol or methanol) NaOH or KOH solution were used for the catalyst recovery with no change in activity when compared with aqueous NaOH or KOH washings.
In a 4-reaction cycle the yields were as follows, respectively, 94, 72, 36, 92% yields. The first used fresh catalyst and was not recycled until after the third reaction showing the effectiveness of said recovery procedure.
Act quickly or not at all.
(Hive Addict)
06-12-03 11:35
No 439559
(Rated as: excellent)
US Patent 3458576
Reduction of Arylnitropropenes
Abstract:
Phenylnitropropenes are hydrogenated under mild conditions, e.g. at a temperature under 100*C and a pressure of less than 75psig., in the presence of a slurry of a catalyst in the hydrogenation solvent which has been treated at 45*C for about 4 minutes prior to the hydrogenation. Pd, Pt, and mixtures of these metals with non-pyrophoric nickel are the catalyst used.
Treatment of the Catalyst:
The catalyst is first slurried in the hydrogenation solvent, e.g. methanol. The actual time and temperature conditions employed may be varied to some extent depending on the boiling point of the solvent and the particular catalyst used. The amount of time required for the heating is indirectly proportional to the amount of heat applied. For the preparation of catalysts for the reduction of the title compounds, it has been found that the best time and temperature are 3-5 minutes and 40-50*C, respectively. After the heat treatment, the solvent mass is cooled with a water bath. The mixture is added to the hydrogenation vessel shortly after its preparation for use.
Example 1:
The materials employed are as follows: 12.5 parts 1-phenyl-2-nitropropene, 53 parts absolute ethanol, 1 part glacial acetic acid, 2.5 parts of 5% Pd/C and enough H2 to maintain a pressure of 64 psig for the duration of the reaction. Temperature ranged from 46-67*C. 9.02g of amphetamine sulfate was recovered from the reaction mixture after isolated using standard acid/base procedures. This corresponds with a yield of 64%.
Notes:
A minimum of 0.01 parts of noble metal catalyst must be applied to the non-pyrophoric nickel catalyst for the benefit of this treatment to work. Pd/C is the preferred catalyst. For unexplained reasons, pure nickel catalysts are unable to receive benefit from this treatment. It is apparent though that the mixtures act much differently than the separate pure catalyst. Operating hydrogenation pressures after treatment range from below 60 to over 500psig. The optimum pressure being 75psig. Operating temperatures after treatment range from 25-100*C. The temperature found to be optimum changes as per solvent used. Methanol is best employed at a temperature of 25-50*C while ethanol is best used from 45-80*C. In most cases, 1.5-2hours times was enough for hydrogenation completion.
Act quickly or not at all.
(Hive Addict)
06-12-03 12:12
No 439574
Low yielding or otherwise not advantageous patents for butyrolactone
US Patent 3113138
82-94% butyrolactone from succinic anhydride
94% butyrolactone from maleic anhydride
(just a primary reference from the procedure given below)
Uses the procedure found in
Post 439554 (Aurelius: "US Patent 3214385 butyrolactone catalyst recovery", Methods Discourse)
US Patent 287628
20% butyrolactone from succinic anhydride
US Patent 2772292
74% butyrolactone from succinic anhydride
(uses 2000+psi and 250*C+)
Act quickly or not at all.
(Heavyweight Chempion(eer))
07-19-03 05:04
No 448417
(Rated as: excellent)
Low-pressure catalytic hydrogenations of nitroalkenes to aminoalkanes have always interested me. Unfortunately it has always been next to impossible according to the litterature unless massive amounts of catalyts are used. Patent US3458576 [pdf] changed this with a great step in the right direction. The authors still use a huge amount of catalyst. But I guess I can live with that.
The method described was tried with 1-(2,4-dimethoxyphenyl)-2-nitropropene but gave only 23% amine. I suspected the low amount of acetic acid used in the patent could be the reason so I tried it again this time with 5 mol eq. acetic acid. This time I got 58% amine. In the trial I used 20% w/w 5% Pd/C pre-treated with 4 bar H2 at 65°C for 6 minutes, for 8g of the nitroalkene in 50ml EtOH. The hydrogenation temperature was then 65-75°C and the pressure was kept between 4-5 bar for 4 hours.
Moody as hell
(Hive Bee)
07-19-03 10:42
No 448463
Old Germans use al lot of Pd/C. In the patent
Patent DE968545
they use 50g Pd/C to convert 60g Ephedrine/Hcl in Meth.
But it works
roger2003
(Hive Bee)
07-19-03 17:52
No 448535
Old Germans use al lot of Pd/C. In the patent
Patent DE968545
they use 50g Pd/C to convert 60g Ephedrine/Hcl in Meth.
But it works
In this patent they use palladium sponge what is not Pd/C at all. Thats plain palladium black in spongeous form AFAIK. The amount palladium is enormous here! I believe the patent belonging to this kind which are made for a company planning to manufacture a compound to proof to own a procedure and so not having to pay license fees. How they really do it in the factory - nobody knows.
But yes, it should work. If a benzylic alcohol can be reduded with palladium this should work.
Don´t mind roger, UF made the same mistake when he transfered this patent in one of his books. ![]() He also forgot to reference it.....
He also forgot to reference it.....
(Hive Bee)
07-20-03 02:06
No 448619
You are right
"Palladiummohr" means fine powdered palladium black
http://www.seilnacht.tuttlingen.com/Lexi
(Hive Bee)
08-03-03 15:56
No 451513
The method described was tried with 1-(2,4-dimethoxyphenyl)-2-nitropropene but gave only 23% amine. I suspected the low amount of acetic acid used in the patent could be the reason so I tried it again this time with 5 mol eq. acetic acid. This time I got 58% amine. In the trial I used 20% w/w 5% Pd/C pre-treated with 4 bar H2 at 65°C for 6 minutes, for 8g of the nitroalkene in 50ml EtOH. The hydrogenation temperature was then 65-75°C and the pressure was kept between 4-5 bar for 4 hours
Barium, will this method work with nitromethane in the conversion to methylamine? The pre-treatment of the catalyst in the acetic acid is for what,... to modify the catalyst ? Is the reaction necessary to heat up since I've herd that most nitrate reductions are exothermic?
In using a Parr Hydrogenator the hydrogenation is fast and efficient, hence so why would it take so many hours to hydrogenate ? Is the hydrogenation of nitrates that difficult for the hydrogen to replace the Oxygen and make H2O and methylamine. I'm willing to try it but want to read all the information and possible hazards noted by those that have tried the procedure.
I missed your post so I started a thread on the search for the answers with someone that has had hands on or is familiar with the literature with the process.....java
Post 451095 (java: "A question about methylamine synthesis", Chemistry Discourse)
(Heavyweight Chempion(eer))
08-04-03 07:16
No 451607
Hi Java, the reaction conditions I used to make 2,4-dimethoxyamphetamine from the nitropropene is not necessary if you want to make methylamine from nitromethane. Beside from not being needed, those conditions would be outright dangerous.
Nitrometane is by far easier to reduce than the nitropropene I used so both lower temperature and a lower catalyst loading would be needed in order not to get a very violent exotherm. I would use about 5% w/w 5%Pd/C (5g catalyst/100g nitromethane) while keeping the reaction temperature at between 40-60°C and the pressure at 3-5 bar.
The pre-treatment I used is jsut a way to "activate" the catalyst. What happens is probably that the catalyst is saturated with hydrogen. Why it gives this effect I don't know.
The Parr-system is outdated nowdays both in terms of safety and efficiency. The bottles used doesn't have the correct shape for work under pressure and the mixing by shaking is not particulary efficient to transfer gaseous hydrogen to the catalyst surface. The reactor should have the form of a cylinder with a rounded bottom (like a gas cylinder). Parr uses flasks with flat bottoms as well as a neck - not good. High efficiency mixing is done with stirring not by shaking. This gives the highest surface area of the gaseous hydrogen which is of utmost importance for a good and steady reaction rate.
Moody as hell
(Hive Bee)
08-04-03 12:22
No 451654
Barium: thanks for the direction as I plan to put a cooling jacket on my parr borosilicate bottle with a thermocuple to monitor the temp . So I can run it in methanol while observing the catalyst ratio your provided , also assuming the pre treatment of the catalyst is not necessary. . While the reaction is exothermic I will cool it down with the water circulating jacket, such that at the end I will have methyl amine in methanol. I could then gas it or add Hcl and remove the methanol to get methylamine Hcl salts , correct me if I'm wrong......java
P.S. thanks again for your help as there is much misinformation about this procedure from the impossible to the extreme danger, but then none had hands on experience to give first hand reports. /td>
(Hive Bee)
08-05-03 04:51
No 451781
(Rated as: good read)
Poisioning and deactivation of palladium catalysts:
Journal of Molecular Catalysis A: Chemical 173 (2001)275-286
http://www2.sivento.de/sivento/uploads_a
roger2003
(Heavyweight Chempion(eer))
08-06-03 05:45
No 452086
(Rated as: good read)
I plan to put a cooling jacket on my parr borosilicate bottle with a thermocuple to monitor the temp . So I can run it in methanol while observing the catalyst ratio your provided , also assuming the pre treatment of the catalyst is not necessary. . While the reaction is exothermic I will cool it down with the water circulating jacket, such that at the end I will have methyl amine in methanol. I could then gas it or add Hcl and remove the methanol to get methylamine Hcl salts
Do small batches first until you get the hang of how this reduction works. Then you can increase the batch size stepwise. What cooling area does your jacket provide? Remember that glass is a poor heat conductor so any strong exotherm might become a runaway reaction if you can't cool it down quick and efficient.
If you run a hydrogenation and the temperature and pressure starts to rise rapidly, the first thing to do is to cut the hydrogen supply and stop the stirring/shaking. This kills the reaction since no more hydrogen is delivered.
Don't start with a 100g batch. Start with a 10g batch and work your way up.
The Unworthy
(Hive Bee)
08-06-03 18:56
No 452208
Barium, thank you again for your insight and safety tips.
Keeping in mind that glass is a poor heat conductor and not having the modern reactor from Parr with the internal cooling coils, but the old type rocker. With no other way to cool but to put a cooling jacket around the glass container, with very little cooling. Not to be discouraged though, it will be attempted in small quantities until a good ratio of concentration of nitromethane and catalyst is found as to keep the reaction under control.
In another page I read foxy2 find using the raney nickle with a yield of 80% of methylamine at room temp in just a few minutes......
Post 353051 (foxy2: "Raney Nickel CTH Reduction of Nitro/Nitrile Groups", Methods Discourse)
this can be a good alternative.......java
(Stranger)
02-29-04 09:29
No 491813
Well, sice you are considering reducing nitromethane to methylamine with Pd/C, this might interest you:
Post 490818 (armageddon: "Reductive amination", Chemistry Discourse)
Or, perhaps more informative, http://www.tech.chem.ethz.ch/hungerb/res
Obviously, calorimetry is VERY important in terms of controlling industrial-scale hydrogenations with Pd/C...
They even designed a reaction calorimeter for that particular purpose!
..better have that bucket of ice/salt-water on hand....
Parr hydrogenators aren't nice when they explode, especially if they contain something like nitromethane!
http://tlf.cx/dearpenis.swf
(Hive Bee)
03-01-04 09:07
No 492016
32 pages Hydrogenation Overview from Degussa:
http://www.strem.com/code/degussakit.htm
"Recommendations Guide"
roger2003
(Stranger)
03-04-04 01:54
No 492915
WOW! That's a pretty cool compilation you found roger2003, THX! And the best things are page 4 (hydrogenation of c=c double bonds) and p. 17 (hydrogenation of alipathic nitro groups)...
According to these reaction schemes, the amount of hydrogen being necessary to achieve complete reduction of P2NP to Phenyl-2-aminopropane would be 8 molar equivalents, right? three H2 for the nitro group and one H2 for the double bond? What if one used ammonium formate as a hydrogen donor, would 8 molar equivalents be sufficient for "complete" reduction of P2NP (as complete as possible under these conditions, maybe 60% of theory? dunno..) or is it better to use even more HCOONH4, SWIMs condenser was severely clogged with white precipitate last time he did a reduction... obviously this was a product of ammonium formate decomposition (maybe ammonium carbonate?), so maybe SWIM should account for that next time and use even more of it...
And wouldn't silica gel help to improve yields, taking up the water formed in the nitro-to-amine hydrogenation (2 mol H2O per mol of styrene)? (Hmm, 120g silica gel for one mol nitrostyrene? it can soak up 30% of its weight of water)
Correct me if my few brain cells worked improperly here..
Oh, and a last question: in their hydrogenation overview, degussa states that the favorable temp. range for nitro reductions is 50-150°C, but the optimum temp. for double bond reductions is 5-100°, so what temp. between 50-100°C would be best for nitropropene reductions? I would think 100°, but I'm just guessing here. Any suggestions?
Thanks!
http://tlf.cx/dearpenis.swf
(Hive Adickt)
03-04-04 12:15
No 492995
You need high presure to reduce the nitropropene to the amine (much more than 3-6bar). Iff that's not posible stick to the two step reation. From nitropropen to nitroprpan and then to the amine (using RaNi (cheap)) Pd/C (starting material must bee pure but yeald is high) CTH(the best) Zn/AcOH(most getto) ect.
(Stranger)
03-04-04 12:48
No 493001
Thanks for your response, hest!! But are you sure about the pressure thing? I'm thinking of
Post 108528 [missing] (dormouse: "reduction of P2NitroPropene with Pd-C -dreamer", Novel Discourse) and
Post 11380 (psyloxy: "on reductions with Pd/C...", Chemistry Discourse)
I thought the procedure worked as described by dreamer.... he obviously didn't use pressure/hydrogen, but used NH4COOH as internal hydrogen source (the formate is decomposed by the Pd/C and releases hydrogen)..
SWIM did some small scale experiments with P2NP a time ago, trying to use ammAcetate/formate as hydrogen source (the latter with more success
Any idea what happened with his P2NP? The yellow oil SURELY was no P2NP at all! So something must have happened to it... Anyone who knows what it could be?
Anyway, how could one reduce said nitropropene to the amine without using NaBH4 (expensive, hard to get) and forming the nitropropane, then reducing the nitro group? Besides of making ketone with HCl/Fe or the like? I'm all ears!
Thanks!
http://tlf.cx/dearpenis.swf
(Stranger)
03-04-04 23:03
No 493102
Since SWIM is a VERY curious person sometimes , after I told him some hive bee stated pressurized hydrogen would bee necessary, he decided to find out if a Pd/C reduction of phenyl-2-nitropropene to phenyl-2-aminopropane (amph.) would bee possible without pressure. He instead wanted to use ammonium formate as hydrogen donor, like described in Post 108528 [missing] (dormouse: "reduction of P2NitroPropene with Pd-C -dreamer", Novel Discourse) with slight modifications: SWIM wanted to use isopropanol as solvent, let the reaction run under anhydrous conditions and he wanted to use 8 mol. equivalents of ammonium formate. So, he reXtalized some P2NP, and got beautiful yellow needles up to 2cm in length...
Five minutes later, SWIM held his breath, removed the condenser and added the 2nd portion NH4COOH (adding it through this condenser would've been impossible
He then added the remaining 2 portions in 10 minutes intervals, and he cranked the hotplate up to ~150°C. After 40 minutes total refluxing time, he cooled everything, removed his now snow-white condenser (perhaps it is not really superb for this purpose, SWIM thought) and added some benzene (no toluene at hand! bad..). Then he did one of the slowest filtrations he ever did...
Vacuum would've been nice here...
...hours later, SWIM added some water and sodium carbonate to his solution, and a yellow benzene layer separated out. Unfortunately, SWIM had broken his 1000ml sepfunnel two days ago, so he had to use a pipette to transfer this yellow layer to a clean flask. The benzene was removed by distillation, leaving a yellow oil which was dissolved in 15% HCl. This acid solution (yellowish, some darker stuff floated on top) was "washed" with colemans
Well, there was still enough oil layer present after his accident, so SWIM separated it with his shitty "new sepfunnel", added colemans, separated, destilled the coleman away and got 2.7ml of clear, yellow oil which was slightly soluble in water (ph ~8.5) and smelled exactly like some kick ass street dope a friend had given him "to check its purity" just a few hours ago (and it WAS pure!)...
SWIM hasn't slept since then.
Maybe because of the vapors he inhaled while he repeatedly checked the smell of his oil, being impressed about how identical to "real" amphetamine it was indeed...
(he smelled it AT LEAST a dozen times, just to be sure...) Not to forget the vapors during his accident..
SWIM told me he dissolved the oil in 99,9% acetone, added a few granules of silica, stoppered it and placed it in the freezer. Now he's waiting for his sulfuric acid to arrive, it's ordered...
But I know NO substance that would have made SWIM wanting to smell it again and again, besides amph. (weed doesn't count, as it is unfortunately no product of the described reaction, not even a side product
Will tell you about SWIMs efforts in getting sulfate salt as soon as he will tell me.
Oh, I forgot: SWIM told me the reason why he first heated to 100°C, then slowly to 150° was he wanted to get first optimal conditions for double bond hydrogenation (50-100°), and then shift to conditions best suited for nitro reduction (100-150°, some more AcOH as additional solvent) Thanks again roger2003!
(am I off-topic? forgive me, but that's the only active Pd/C thread..)
(Pioneer Researcher)
03-05-04 04:13
No 493130
If you add formic acid instead of acetic you'll get an extra portion of am. formate. Your supposed amphetamine is actually amphetamine isopropyl imine... It have reacted with your anhydrous acetone. You shoud acidify in aqueus medium to go back to amphetamine, then extract and make the same but with ether, toluene etc...
(Stranger)
03-05-04 19:12
No 493320
Thanks for your response, sunlight! Well, SWIM told me he added the GAA/60%acetic because the very complete hydrogenation overview at http://www2.sivento.de/sivento/uploads_a
Hydrogenation of Aliphatic Nitro Groups
R-NO2 H2 > R-NH2
Precious metal: Pt, Pd
Conditions
Temperature range [�C] 50-150
Pressure Range [bar] 3-50
Supports activated carbon
Solvents low polar solvents, acetic acid
Catalyst modifier e.g. Fe, V
But SWIM thinks that adding some additional formic acid won't hurt, but will reduce the amount of NH4COOH needed, thus also reducing the amount of white precipitate in his shiny, but difficult to clean ![]() condenser. but he thinks he will use acetic, too next time. But why "amphetamine isopropyl imine"? The yellow oil sitting in SWIMs freezer was the product of a/b extraction, followed by removal of the nonpolar through distillation - there was very little chance that there was isopropyl alcohol left from reaction. Besides, I see no reason in acidifying/extracting a freebase acetone solution that was already acidified in aequous medium, then basified and extracted w/nonpolar which was removed..
condenser. but he thinks he will use acetic, too next time. But why "amphetamine isopropyl imine"? The yellow oil sitting in SWIMs freezer was the product of a/b extraction, followed by removal of the nonpolar through distillation - there was very little chance that there was isopropyl alcohol left from reaction. Besides, I see no reason in acidifying/extracting a freebase acetone solution that was already acidified in aequous medium, then basified and extracted w/nonpolar which was removed..![]()
And, sorry to question your statement, dear sunlight, but should SWIM really try to extract an acetone solution with a non-polar (no matter whether it is basified or acidified bee4)? I think the acetone would really fuck up the two phases, and make further separation impossible. You surely meant SWIM should remove the acetone by distillation, dissolve the remaining oil in dil. HCl, basify and THEN Xtract w/nonpolar, hm? But anyway, SWIM will try to get the sulfate like planned as soon as his sulfuric arrives, of course he will make up a solution of it in acetone and drip that into his yellow oil/acetone mix, if no precipitate appears, you're probably right about amph isopropyl imine formation. But in that case SWIM could add just H2O, some more acid, distill away the acetone, basify, extract and try again with benzene as solvent, right?
Sunlight, just for better understanding, could you explain how said imine would be formed if that was the case? I'd appreciate it very much! (dreamer claimed having formed the sulfate in acetone and ether in this thread Post 108528 (dormouse: "reduction of P2NitroPropene with Pd-C -dreamer", Novel Discourse) and SWIM as well as me thought the ether was just left over from extracting, and acetone did the trick - due to the solubility of sulfate being very small compared to fairly high solubility of the free base..)
corrections are always welcome!
THX armageddon![]()
(Stranger)
03-05-04 21:18
No 493343
Since this is the Pd/C thread...
Check out http://www2.sivento.de/sivento/uploads_a
...and Patent US6096924 [pdf] about the use of vanadium as hydroxylamine disproportionating catalyst (unfortunately they reduced only aromatic nitro compounds, but the mechanism behind it is interesting anyway, perhaps a new yield booster for nitro reductions? Note the disproportionation is a shortcut, it significantly reduces the amount of hydrogen needed for total reduction, as it avoids the azo route)
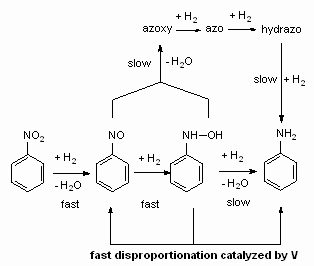
Hope this will help some bees in understanding Pd/C better...
(Stranger)
03-06-04 08:54
No 493438
Well, Sunlight, of course you were right and SWIM was wrong... ..you deserve it well being called "pioneer researcher"! And adding water to the acetone-"solution" produced 2 layers on acidification! DOH, SWIM thought, same thing as with post-rxn-mxtr.; alcohol is soluble in both polar and nonpolar solvents, but the addition of water causes separation of NP and alcohol..
One more error to correct: Dreamer used ether/ethanol, not acetone for precipitating the sulfate, the (obviously not working) procedure SWIM tried to use without success is in the "urushibara style amphetamine" document on Rhod's page... Unfortunately, SWIM got < 1ml when he acidified (having first added some H2O) and basified/extracted/distilled again, and it still didn't form any precipitate (solvent: dry toluene this time). Well, SWIM thinks that some acetone has made it through the A/B process, again interfering with precipitation
At the moment, SWIM is making more P2NP to be able to try again what smelled really as if it was successful
He's SURE the complete reduction in one step works, and more experiments will be made. Next time, adding formic acid is on the to-do list along with using less formate, using strictly lab grade solvents and reagents (NO colemans
If someone has ideas about what else could bee made better, tell me!!
THX a
(Stranger)
03-10-04 22:56
No 494322
Since his last experiment wasn't very successfull, SWIM decided to try once again to reduce P2NP to amphetamine directly via Pd/C and ammonium formate as hydrogen donor. Not wanting to waste any more of his P2nP (it was his 3rd try with this route
First, SWIM weighed out 4,4 grams of Pd/C (10% Pd, used one time and reactivated like described in Patent US3214385. [pdf]
The Pd/C was put in a 500ml RBF, moistened with iPrOH, then 40 ml AcOH (last try was with some formic and mainly produced loads of CO2 smoking from top of his condenser
SWIM then heated to reflux (ca. 100°C). As soon as reflux started, he introduced 3x 13g NH4COOH every 9 minutes and one last extra 5g portion was introduced after 25 minutes. After 35 minutes, SWIM cooled everything, added ~100ml DCM and set up for gravity filtration. (AAARGH!! It took 3,5 hours!
Then he added water until the DCM separated out nicely (~500ml). The DCM was separated, distilled away and SWIM got 11,7ml of a yellow oil with a VERY distinctive smell
Because SWIM always wants to be accurate, he decided to carefully distill away the toluene for being able to determine the amount of freebase, but unfortunately he ended up with just 3,3ml of a clear, yellow oil, pH of aequous soln. was 5-6 and it dissolved it HCl... and had THE SMELL..
Since his last attempts in forming the sulfate had failed miserably (got nothing..), he tried to be very careful this time. He thought "Hm, for a 4M solution, I would need to dilute 4,93ml of my oil to 10ml with iPrOH, assuming it was pure amine freebase. Ok, some toluene may be still in it, so I will use 0,6ml and dilute to 1ml and assume it is 4M concentration. And the sulfate is (C9H11NH2)2*H2SO4, so I should be able to neutralize my 4M with an equal amount of 2M sulfuric/iPrOH, or at least not over-acidify everything". Said, done. SWIM diluted his oil in a test tube, added calculated amount of acid soln. - CLEAR YELLOW...
But he only got 150mg of white (very pure) sulfate!
Perhaps I should have dried my solvents with some drying agent, SWIM thought disappointed as he was drying his crystals. Or perhaps I should'nt have distilled off the toulene, perhaps there was still water present, and the freebase was steam distilled, but then, crystal formation should've been easier (nearly anhydrous conditions)
Can anybody help SWIM in improving his shitty yields (1%!?
He would greatly appreciate it!
Thanks A
(Hive Bee)
05-09-04 04:48
No 505982
Note to myself: Sulfuric acid dissolved in alcohol is incompatible with silica gel - if you let it stand several days the silica gel will partially dissolve into something very viscous and stinky - perhaps Si(SO4)2 or something similar. At least the sulfuric solution becomes unusable...
"..ein Trank von unterschiedlicher Farbe, in ihm ist Heilung für die Menschen."