(Chief Bee)
09-11-01 14:13
No 212038
(Rated as: excellent)
This post was edited to resolve stereoconfiguration confusion on September 3, 2002. /Rhodium
After reading J Chem Soc, 850-854 (1952), I found ideas for 4-Methylaminorex (4-MAR, U4EUh) syntheses, with or without the use of cyanogen bromide. All the examples below relates to the reaction of ephedrine and pseudoephedrine to form racemic 3,4-dimethylaminorex isomers, but the exactly same schemes should apply to phenylpropanolamine (PPA) to give 4-MAR. In this text, the designation phenylpropanolamine encompasses both norephedrine and norpseudoephedrine and their respective optical isomers. The two routes described are 1) The classic one-step cyanogen bromide cyclization of PPA and 2) Formation of the carbamyl (urea) derivative of PPA using potassium cyanate, followed by acid cyclization.
Using the cyanogen bromide route, norephedrine (1R,2S)/(1S,2R) yields cis-4-methylaminorex and norpseudoephedrine (1S,2S)/(1R,2R) yields trans-4-methylaminorex. The cyanogen bromide procedures below aren't optimized, addition of 3 equivalents of sodium acetate and doubling the amount of cyanogen bromide would not produce a precipitation of ephedrine salts, thus making the procedure more effective (see other syntheses of 4-MAR on my site). Also note that cyanogen bromide is very toxic.
Using the "double racemic" phenylpropanolamine (1RS,2RS) would give equal amounts of racemic cis- and trans-4-methylaminorex. All the cis/trans isomers are active, as well as their respective stereoisomers. The trans(4S,5S) is the most potent, with an effective dose of 0.25 mg/kg (compared to dextroamphetamine considered active at 0.4 mg/kg and d-meth at 0.2 mg/kg). The cis isomers are of about 5 times lesser potency, but they are still pretty active, as they are about equipotent to racemic amphetamine, and with a 2-3 times longer duration.
Using the potassium cyanate route, it should be noted that norephedrine (1R,2S)/(1S,2R) will be transformed into trans-4-MAR (the opposite of the cyanogen bromide route), while norpseudoephedrine (1S,2S)/(1R,2R) upon the same treatment instead forms trans-4-methyl-5-phenyl-oxazolid-2-one (which looks just like 4-MAR, but with a double bonded oxygen instead of the NH2 in the 2-position). It is an amide rather than an amine, so it should be removable using an acid-base extraction. Thus, no cis-4-MAR can be produced using potassium cyanate.
The byproduct amide trans-4-methyl-5-phenyl-oxazolid-2-one can be isolated and catalytically hydrogenated at room temperature in ethanol containing 5% triethylamine and 10 mol% Pd/C to form (S)-amphetamine in 90% yield, Ref: Chem Eur J, 3(8) 1370 (1997), but that is probably not particularly useful, but using the potassium cyanate scheme on substituted (nor)ephedrines (like the 3,4-methylenedioxy variety) would enable you to produce a stereoselective synthesis of (S)-MDMA, the more active isomer, together with the 3,4-methylenedioxy-trans-4-MAR for evaluation of its activity. Edit: The article can be found in Post 421261 (Rhodium: "Stereoselective (S)-MDA synth via the Cyanohydrin", Novel Discourse)

Experimental
N-Carbamyl-(±)-ephedrine
To 5g (±)-Ephedrine hydrochloride (25 mmol) in 25ml water was added 2g potassium cyanate (KOCN, 25 mmol), and the solution was heated under reflux for 2.5 hours, during which time a small amount of oil separated, then the solution was cooled in an ice-salt bath. The dried, white plates of the formed urea (3g, 57.7%) were recrystallized from ethyl acetate and was found to have a mp of 126-127°C.
(±)-trans-3,4-Dimethylaminorex HCl
A solution of N-carbamyl-(±)-ephedrine (1.56g, 7.5 mmol) in 24ml water and 15ml 2N HCl was refluxed for three hours, when the clear solution was cooled the (±)-trans-3,4-Dimethylaminorex hydrochloride precipitated. This was purified by basifying the solution, extracting it with benzene (can use any non-polar solvent here), and the solvent evaporated and the freebase converted to the hydrochloride by gassing with dry HCl in ether. Yield 1.9g, 84%, mp 225-229°C.
(±)-cis-3,4-Dimethylaminorex HCl (from (±)-ephedrine)
60ml of an etheral solution containing 3.5g (30 mmol) cyanogen bromide was added to 200ml of an etheral solution containing 11g (±)-ephedrine (66 mmol), whereupon 8.1g of ephedrine hydrobromide separated (50% based on ephedrine input, 33 mmol, mp 186-188°C) and was filtered off and washed with ether. The filtrate was concentrated to 25ml, and white needles (1.5g, mp 71-73°C) of (±)-cis-3,4-Dimethylaminorex freebase precipitated. The filtrate was concentrated further, and the residual oil treated with ethanolic hydrogen chloride. The product was recrystallized from a mixture of 25ml CHCl3, 10 ml acetone and 5ml ether yielding 4.2g of (±)-cis-3,4-Dimethylaminorex hydrochloride, mp 215-217°C.
(±)-trans-3,4-Dimethylaminorex HCl (from (±)-pseudoephedrine)
40 ml of an etheral solution of 1.75g cyanogen bromide (16.5 mmol) was added to a solution of 5.5g (±)-pseudoephedrine (33 mmol) in 100ml ether and 80ml benzene. In addition to precipitated pseudoephedrine hydrochloride (3.9g), (±)-cis-3,4-Dimethylaminorex hydrochloride (2.2g) was obtained upon treatment of the residual oil after evaporation of the solvent with ethanolic hydrogen chloride, mp 215-217°C, identical to the sample prepared above.
(Stranger)
09-11-01 15:14
No 212048
The use of cyanate certainly makes synthesis of these compounds in an informal atmosphere more feasible. Cyanogen bromide really shouldn't be played with without proper equipment.
Still wondering what effects that ring substituted aminorexs have (e.g. 3,4-methylenedioxy aminorex). Pihkal gives the lead on how to make the starting materials. For instance, reaction of piperonal with sodium cyanide to give the cyanohydrin, followed by LAH reduction to give the amino-alcohol.
(Distinctive Doe)
09-11-01 20:44
No 212179
Anyone have any idea about the activity of 3,4-Dimethylaminorex HCl?
Do Your Part To Win The War
(Chief Bee)
09-11-01 21:06
No 212184
According to this page, it may not be healthy: http://www.biopsychiatry.com/4-methylami
(Distinctive Doe)
09-11-01 21:34
No 212192
Well depending on the numbering scheme used I belive the methyl in the compount they mention is on the benzylic carbon and not the nitrogen. They would call it an N-methyl.
Here is the quote about neurotoxicity.
"Aminorex and its analogues, with exception of 4S, 5S-dimethylaminorex, did not cause the long-term neurotransmitter depletion in either the dopaminergic or 5-HT-ergic systems."
Euphoria(4-Methylaminorex) according to chemfinder is this 2-amino-4-methyl-5-phenyl-2-oxazoline.
So it appears to me the product from ephedrine is NOT what they are talking about. That is assumeing the numbering scheme is the same, however I am having a hard time figuring out the logic behind the numbering. It would be good to get the article to see for sure.
Do Your Part To Win The War
(Chief Bee)
09-11-01 23:49
No 212238
The "4S, 5S" thing is to indicate the chirality of the carbon atoms at the 4- and 5-position on the heterocyclic ring, they don't indicate the position of the second methyl. It could be anywhere, but I find it most likely it would be on the 3-positioned nitrogen if nothing else is said. But I guess someone have to fetch that article just to be sure.
(Stranger)
09-13-01 10:20
No 212997
This seems to be exactly what I was looking for Chief.
It looks to me like your new method will also work for NorPseudoE.HCL HaHaHaHaHa!
How come I'm seeing HOCN in the pictures and KOCN in the literature. Which one did you edit Mr. Chief Bee?
See you guys on the other side.......
(Chief Bee)
09-13-01 13:02
No 213026
The reaction calls for pseudoephedrine hydrochloride and KOCN, which in my reaction diagram (and the original diagram in the article) has been "translated" to HOCN and pseudoephedrine freebase. I consider the terms to be interchangeable, because the intermediate product (N-carbamyl-(±)-pseudoephedrine) is essentially an addition of HOCN to pseudoephedrine if you count the atoms. The Cl- from the hydrochloride and the K+ from the potassium cyanate are only spectator ions in the reaction.
To my knowledge, free HOCN cannot be made as a stable solution, that's why it is generated in situ.
(Newbee)
09-13-01 13:52
No 213044
Thanks for that little clarification Chief!
I still say that logically d-NorPseudoEphedrine will work and the beauty of it is that you then, according to your refs above, would end up with the trans isomer of 4-MAR.
4-DMAR would be produced with PseudoEphedrine.
Nor, Nor, Nor. it's the Nor that makes the difference boys.
Let the flames begin!
(Chief Bee)
09-13-01 14:38
No 213050
I would welcome any reports of using norpseudoephedrine (phenylpropanolamine) in the potassium cyanate reaction, either in this thread or in email.
Now when we know that the reaction proceeds through an urea derivative, what other methods are at our disposal to form it? Reaction of urea with a styrene halohydrin?
(Newbee)
09-14-01 12:03
No 213453
This sound good Rhodium, this bee is loaded with Urea. Not so sure about the styrene halohydrin though. I am rather pissed with Halogens in general after my dismal failure with that DMSO/NaI/H2SO4. I produced more tar than anything with a final yield of 14%. Cleanup sucked as there was I2 everywhere and let's not even talk about cleaning glassware after a tar invasion.
Do you have any refs or procedural material for this?
(Hive Addict)
09-14-01 20:40
No 213584
Wow this looks to be an easy synthesis!
(Newbee)
09-15-01 11:38
No 213811
Rhodium, correct me if I'm wrong, but isn't HOCN cyanuric acid?
'Cause if it is...... there's lots to be had at our local pool store. hint hint
(Chief Bee)
09-15-01 16:22
No 213833
I believe the pool store thing is a trimer of HOCN (don't know if it can be depolymerized readily), but I may be wrong, as I have no reference literature with me where I am now.
(Distinctive Doe)
09-16-01 01:47
No 213910
Soon I will post some easy routes to the salts of HOCN.
Very easy, I just cannot access the stuff over the weekend.
Hold on til monday
Do Your Part To Win The War
(Hive Bee)
09-16-01 02:12
No 213914
I am positive that addition of water with depolymerize the cyanuric acid.
(Distinctive Doe)
09-16-01 20:47
No 214057
(Rated as: excellent)
United States Patent 4,000,249
Sochol , et al. December 28, 1976
Preparation of alkali metal cyanates
Abstract
Process for the preparation of alkali metal cyanate by the reaction of urea and alkali metal carbonate. The addition of water to the reaction in an intermediate step activates partially blocked alkali metal carbonate and provides an alkali metal cyanate product of high purity.
Here are some other patents
Sodium cyanate from urea and sodium. Besson, Paul; Aigueperse, Jean. (Ugine Kuhlmann). Fr. (1972), 6 pp. CODEN: FRXXAK FR 2110788 19720707 Patent written in French. Application: FR 70-39205 19701030. CAN 78:60320 AN 1973:60320 CAPLUS
Abstract
Na is dissolved in an alc. having a b.p. between 110 and 160°. Urea is also dissolved in the same alc. and heated to boiling. An equimol. amt. of the Na alcoholate soln. is added to the urea progressively over a period of ł30 min and the mixt. is left to react until no more NH3 separates. The cryst. product is very pure NaOCN which is substantially free of Na2CO3 and NaCN. The alc. is preferably EtOCH2CH2OH.
Alkali cyanate. Iwai, Tomiro. (Japan Fine Chemicals Co., Ltd.). Japan. (1971), 4 pp. CODEN: JAXXAD JP 46040536 19711130 Showa. Patent written in Japanese. Application: JP 19610509. CAN 76:87988 AN 1972:87988 CAPLUS
Abstract
All the Na2CO3 to be reacted with urea is heated to 150-350° at first. To this dry powder, urea is added little by little with mixing by stirring. The heating is reduced gradually as the reaction proceeds. High-purity alkali cyanate is then obtained with a yield of 96-9%. Alkali cyanide is undetectable in the product.
Sodium cyanate from urea and sodium carbonate. Schunk, Wolfgang; Hohn, Richard; Jasche, Klaus; Braumann, Dietrich; Schweizer, Heidrun. (VEB Agrochemie Piesteritz, Ger. Dem. Rep.). Ger. (East) (1985), 8 pp. CODEN: GEXXA8 DD 221449 A1 19850424 Patent written in German. Application: DD 84-260213 19840221. CAN 103:162702 AN 1985:562702 CAPLUS
Abstract
High-quality Na cyanate free of CN- and of reactor corrosion products is obtained in 3 sequential stages from urea and Na2CO3 at mol ratio (1.8-2.2):1 and Ł180°. A mixt. of Na allophanate and Na cyanate is formed in the 1st stage at 100-120°, water is evapd. in the 2nd stage at 130-140°, and Na allophanate is converted into Na cyanate in the 3rd stage at 140-180°, the reaction gases being simultaneously removed in all stages. Thus, 471 wt. parts of 95.6% Na cyanate was prepd. from 424 wt. parts of Na2CO3 and 480 wt. parts of urea.
Sodium cyanate. Dragalov, V. V.; Karachinskii, S. V.; Chimishkyan, A. L.; Izakson, G. Yu.; Shvets, P. K.; Kulygin, A. A.; Kulygina, A. F.; Turlanov, N. N. (Mendeleev, D. I., Chemical-Technological Institute, Moscow, USSR). U.S.S.R. (1984), CODEN: URXXAF SU 1074818 A1 19840223 Patent written in Russian. Application: SU 82-3513458 19820917. CAN 100:158962 AN 1984:158962 CAPLUS
Abstract
NaOCN is manufd. by reacting urea and Na2CO3 at 125-170° with mixing and addn. of portions of urea to the Na2CO3. The yield of the product is increased and content of a basic substance in the product is increased by cooling the reaction mixt. to Ł20° prior to addn. of each successive portion of urea and the resulting NaOCN is held at 180-300° until the gasification of impurities ceases. Urea is added to the Na2CO3 in 3 equal portions.
Manufacture of alkali-metal cyanates. (du Pont de Nemours, E. I., and Co.). Brit. (1969), 5 pp. CODEN: BRXXAA GB 1145777 19690319 Patent written in English. Application: GB Priority: US 19660801 - 19670710. CAN 70:116709 AN 1969:116709 CAPLUS
Abstract
Na cyanate (I) or K cyanate (II) of 98% or higher purity, free of cyanide is obtained by the low temp. reaction of stoichiometric amts. of tech. grade alkali metal carbonate and shotted urea in the presence of a suitable quantity of a heel of 98% purity (45 and 55 wt. % of the heel for manuf. of I and II is of the reaction mixt., resp.) in muller-type mixers or screw conveyors having provision for intensive mixing and rubbing and a suitably efficient heat transfer arrangement. A small excess of urea (.apprx.2%) is desirable to eliminate contamination from unreacted alkali metal carbonate. The low operating temps. employed in the process (95-150° and 95-225° being the optimum ranges for I and II, resp.) do not cause severe corrosive conditions and permit the use of cast iron, wrought iron, or stainless steel reaction vessels and batch or continuous operations are practicable. Heating times of 4-7 hrs., depending on mixing and heat transfer characteristics are required for completion of the reaction. Cyanide impurity detected in the end product is <1 ppm. Thus, charge a jacketed, stainless steel, double-arm mixer, heated with steam at 110 psig. (173°) with 5.20 parts of heel of I of 98% purity. Engage the blades turning at 50 rpm. and heat for 1.8 hrs. to 164°. Add 2.25 parts soda ash and 8 min. later 2.25 parts of shotted urea. Heat the slightly wet and lumpy charge for 6 hrs. After 2 hrs., the batch becomes free-flowing and dusty. Vent off the discharge gases throughout the heating period. The yield obtained of 7.8 parts I from the mixer is 99.5% based on the starting amt. of urea, with a purity of 98% and shows <1 ppm. cyanide. About 86% I is formed during the 1st hr. while the temp. of the mix reaches 140° and 94.9% of the total product synthesized is formed in 2 hrs.
High-purity sodium cyanates. Hottai, Yoshitaka; Narisawa, Kenzo. (Tohoku Hiroyo K. K.). Japan. (1970), 3 pp. CODEN: JAXXAD JP 45002655 19700129 Showa. Patent written in Japanese. Application: JP 19650531. CAN 72:91835 AN 1970:91835 CAPLUS
Abstract
Mixts. (1:1.7-2.0 mole ratio) of Na2CO3 (<250 m grain size) and urea are melted at 110-150° and caused to react to a solid state, and the solid product obtained is finally caused to react at 160-250°. For example, mixts. (1:1.8 and 1:1.9) of Na2CO3 (100 m) and urea powders are put in an alumite bowl, heated at 110° under stirring in an oil bath, the product put in a stainless steel dish, and calcined for 30 min at 160° to give 89.5 and 93.7% NaOCN contg. no NaCN, while 30 min heating at 600° of a control mixt. (1:2.2 mole ratio) gives 81.2% NaOCN contg. 0.8% NaCN.
Do Your Part To Win The War
(Newbee)
09-17-01 09:58
No 214272
Thanks fox!!
This is going to make life a lot easier for all of us.
Steps to victory over the D. the E. and the A.
1. Get your ass over to the drug store.
2. Ask for some diet pill (Can't say which) containing d-NorPseudoEphedrine.HCL
3. Get yourself 500g Urea & 1kg Wash Soda (Na2Co3) (shouldn't arouse any sus...)
4. Go make ICE at Home.
5. Don't use too much of your own product or you may only be able to persue your career after coming back from rehab.
6. Be careful and enjoy Beez!!
7. Don't forget to thank the bigger beez for their efforts!!
(Hive Bee)
09-21-01 21:07
No 216006
WHat are some properties of NaOCN, or KOCN? I beleive that addition of a sodium base to cyanuric acid will form the NaOCN that is desired, BUT I sure would like to kow for sure before anyone tries this...
(Distinctive Doe)
09-21-01 22:06
No 216020
Jim
Is sodium carbonate and urea to difficult for ya??
Do Your Part To Win The War
(Hive Bee)
09-22-01 02:24
No 216083
Urea is located @ chem stores right? I would like to have completely OTC. How easy is pool acid, cyanuric acid, and lye. Really give me a break it wouldn't even involve anything other than addition....
(Master Searcher)
09-22-01 02:46
No 216086
I bought urea a couple times from a plant nursery. I've been using it in a fertilizer mixture that I make up which also includes triple superphosphate (monocalcium phosphate), potassium sulfate, Epson salts, and a mixture containing trace elements comprising boric acid, copper sulfate, iron sulfate, manganese sulfate, sodium molybdate and zinc sulfate.
http://www.geocities.com/dritte123/PSPF.
(Distinctive Doe)
09-23-01 01:51
No 216255
Hey all
Here is i think the lowdown on the steroisomerism shit.
I have the ref rhodium posted.
And he is correct about dimethyl being the compound with a n methyl group as ephedrine. How ever the steroisomerism should be (R,S) with ephedrine and (R,R) with pseudo, I THINK.
However I would make very sure before tasteing this compound. Because the (S,S)dimethyl aminorex can easily produce fatal seziures, the article says!!! Would pseudoephedrine, which is (1R,2R)-(-)-Pseudoephedrine, produce the (S,S)dimethyl aminorex or (R,R)dimethyl aminorex????
In other words I would maybe only try this with ephedrine since it is (1R,2S)-(-)-Ephedrine and would almost surely produce a (R,S)dimethyl aminorex.
Anyone a sterochemisty wizz?
left handed or right handed and all that bullshit
Do Your Part To Win The War
(Hive Bee)
09-24-01 05:12
No 216512
There are two types of instant one time use cold packs. One contains ammonium nitrate and the the other contains urea. The brand containing urea can be found at a popular chain store. Hint: bulls eye.
(Hive Bee)
10-04-01 13:13
No 220200
I've found on Beilstein these US Patents, but I cannot open it on uspto.gov:
3278382 and 3161650
(Chief Bee)
10-04-01 14:29
No 220209
They can be opened using the patent search engine at the bottom of ../rhodium /index.h
(Hive Bee)
10-11-01 11:54
No 223108
So swim made NaOCN from 41g Urea and 102g Na2CO3. Now I have this solid white mass and I'mm not sure how to go about purificaation.
Any beez have ideas? I'll do a complete writeup from NaOCN through the Carbamyl Ephedrine to 3,4-DimethylAminorex with all detail / observations included. I also have some interresting ideas on using PseudoPPA.HCL (d-NorPseudoEphedrine) to make ICE (4-MA) with the same process.
Hold thumbs on this one beez, and thanks to Rhodium for this goldmine novel idea.
(Hive Bee)
10-11-01 15:12
No 223151
So is the PPA found in OTC Sinutab etc. d-PPA, the one that will give 4-MA with the KCNO method?
(Hive Bee)
10-11-01 15:42
No 223156
N-carbamyl-(±)-pseudoephedrine
5g (±)-Pseudoephedrine hydrochloride (25 mmol) in 50ml methanol was added a solution of 2g potassium cyanate (KOCN, 25 mmol) in 150ml methanol, and the solution was heated under reflux for one hour. Potassium chloride was filtered off and the filtrate evaporated to dryness. The residue was recrystallized from 20ml of ethyl acetate to give 2.2g of the substituted urea as white plates, mp 140-141°C.
(±)-trans-3,4-Dimethylaminorex HCl
A solution of N-carbamyl-(±)-pseudoephedrine (1.65g, 75 mmol) in 24ml water and 15ml 2N HCl was refluxed for three hours, when the clear solution was cooled the (±)-trans-3,4-Dimethylaminorex hydrochloride precipitated. This was purified by basifying the solution, extracting it with benzene (can use any non-polar solvent here), and the solvent evaporated and the freebase converted to the hydrochloride by gassing with dry HCl in ether. Yield 1.9g, 84%, mp 225-229°C.
Surely if 25mmol pseudephedrine.HCl = 5g, 75mmol N-carbamylpseudoephedrine cannot be 1.65g?
(Chief Bee)
10-11-01 16:13
No 223165
From Purification Of Laboratory Chemicals, 4th Ed:
Sodium Cyanate, NaOCN, mp 550°C. Colorless needles from ethanol. Solubility is 0.22g per 100g EtOH at 0°C. Soluble in H2O, but can be recrystallized from small volumes of it.
(Hive Bee)
10-11-01 17:41
No 223201
"Urea is located @ chem stores right? I would like to have completely OTC."
You would like to have completely OTC urea?!
Honestly! You can't get any more OTC than that! I used to have an old hardbound laboratory manual around here somewhere, circa 1940's, that detailed how to prepare urea from human urine. Unfortunately, I can't find it. I'm sure the laboratory intructors of the time were amused by that experiment. Part of the procedure involved collecting the "first urine of the morning" and boiling it down into a "syrup".
Mr. Smith recommends eating steak or other high protein food before attempting this procedure.
(Hive Bee)
10-11-01 17:47
No 223205
For those who are curious:
Gattermann&Wieland 'Die Praxis des organischen Chemikers' prepare the KOCN by mixing Kaliumhexacyanoferrat (Fe(CN)6K4) and Kaliumbichromate and dropping spoon by spoon this mixture on a hot iron plate. The KCNO is extracted with boiling alcohol from the resulting rxn mix.
Carpe Diem
(Chief Bee)
10-11-01 18:17
No 223213
Uemura: Is that from the 1850's?
(Hive Bee)
10-11-01 18:36
No 223222
So in the final step should one use 1.65g or 25mmol (5.16g) or 75mmol (15.47g) of N-carbamyl-(±)-pseudoephedrine?
Am I correct in thinking the commonly available OTC form of PPA is d-pseudophenylpropanolamine, the one which by the KCNO method would turn into trans-4-methylaminorex?
Many thankx,
IudexK
(Chief Bee)
10-11-01 19:00
No 223226
The original reference reads 1.56g, 0.075 mole. (seems that I made a mistake in the typing, but it still doesn't make sense).
If 0.0075 moles (7.5 mmol) were used, then the N-carbamoylephedrine would weigh in at 1.56g.
So I typed the weight wrong, and they lost a zero on the number of moles they used.
What is the full stereoconfiguration of OTC PPA? The d-PPA designation does not tell us which way the oxygen points. But only trans-4-methylaminorex can be formed using the cyanate route, if the wrong isomer is used, you will get an amide rather than an amine.
(Hive Bee)
10-11-01 19:42
No 223238
Thankx, I will try and find out what OTC PPA's stereoconfiguration is... but the easiest way would probably bee to try it.
Would [1RS,2RS]-phenylpropanolamine used in the KCNO method give equal parts cis/trans-4-MA?
(Chief Bee)
10-11-01 21:27
No 223262
Racemic norpseudoephedrine would give an inactive amide (removable by an acid/base extraction), and racemic norephedrine would give racemic trans-4-MAR. [1RS,2RS]-phenylpropanolamine (an equimolar mixture of the above two) would give half racemic trans-4-MAR and half the inactive amide.
It does not seem to be possible to create the less active cis-4-MAR using the cyanate route.
Edited to fit the changed stereochemistry in the first post in this thread.
(Hive Bee)
10-11-01 21:41
No 223268
ok, last question, I promise
Do you happen to know the stereoconfiguration of the PPA produced by this synth?: ../rhodium /ppa.htm
Many thankx,
IudexK
(Chief Bee)
10-11-01 21:59
No 223277
It will produce the "double racemic" [1RS,2RS]-phenylpropanolamine.
(Hive Bee)
10-12-01 14:43
No 223631
It's available from my local hardware store. Walk in, buy urea in 25 kg tubs. Ain't NOTHING more OTC than that. Except that it's not over the counter, cause there isn't even a counter, it's OTS - off the shelf!
[red]W101 should listen to his own advice, yes?
In South Africa Urea is in hardware stores.
I don't imagine it makes much difference in this case, but one should practise what one preaches.
Got a surprise there when I saw the melting point - I remembered my ref. saying something about 300 something - went and looked it up, and it said >300 C - I need a better ref. manual
(Junior Service Representative)
10-12-01 19:00
No 223672
Yep, Mr.Smith, I got the same synthesis, but it appears only 4-6g yield from 500ml of urine.
Wonder if the Professors used to skip this experiment...
PB
(Hive Addict)
10-12-01 22:26
No 223737
If I'm not mistaken, all PPA containing products got pulled off of US shelves about a year ago due to FDA claims of "overdoses" - like 14 over a 30 year span. and considering it was in slim fast, I imagine it got abused pretty heavily. your best bet would probably be to extract it out of the ephedra pills containing multiple alkaloids.
Sed quis custodiet ipsos custodes?
(Hive Bee)
10-12-01 22:32
No 223743
I am not in the US, and besides, there are always international pharmacies!
(Hive Bee)
10-13-01 21:57
No 224081
"Yep, Mr.Smith, I got the same synthesis, but it appears only 4-6g yield from 500ml of urine."
Stockpile..... stockpile.....
"Daddy, what's it going to be like in the year 2000?"
(Hive Bee)
10-15-01 22:11
No 224883
To the Chief Bee. You wanted to know: Uemura: Is that from the 1850's?
Ahh, it's more 1950, some kind of post WW-II chemistry
(Hive Bee)
10-17-01 05:46
No 225556
Rhodium:
I don't understand why everyone is afraid of cyanogen bromide. Especially when it can be made in situ from bromine, methanol, and sodium cyanide. All of these items are OTC except the bromine which can be made EASILY by reacting sodium bromide and OXONE in water. The bromine forms immediately without the need for external heat. Since it distills at about 65 deg C or so, it can be distilled at atmospheric pressure directly into the receiving flask (immersed in ice) containing the correct amount of methanol. As for the exit to the vacuum adapter one could lead a tube--with one of those fish aquarium anti-suck-back valves inbetween--directly into a flask containing aqueous sodium carbonate (bicarbonate causes too much foaming) to aborb any gaseous bromine that might escape. And voila! A methanol solution of bromine.
Next, one would dissolve the correct amount of sodium cyanide in methanol and add it via addition funnel dropwise to the ice-cold methanol-bromine solution contained in a flask (attached to a reflux condenser and sitting in an ice bath). Proceed from then on out as per usual.
SWIM once tried this under crude conditions in a open beaker with a blowing fan as a ventillation source. As the sodium cyanide hit the methanol-bromine solution, the bromine color faded and then eventually, it disappeared. Occassionally, there was a faint mist that would form above the methanol solution, but only for a minute.
As for the precursor amino alcohols, they are easily formed from the intermediate nitro alcohols.
According to one paper that I have, Nitro alcohols are formed by condensation of a benzaldehyde and a nitro alkane in aqueous alcoholic sodium hydroxide solution. The nitro alcohols are then reduced in a facile reaction using Zn and aqueous sulfuric acid. Yields are good for one experiment which details the synthesis of phenylpropanolamine. Don't remember the yields but I believe it is somewhere around 80%. Furthermore, I have a reference (CA) wherein MD-phenylpropanolamine is synthesized via the same route as above in over 75% yield.
The cool thing about all this is that phenylethanolamine could EASILY be synthesized using benzaldehyde and nitromethane and Zn and aqueous H2SO4 as the reducing mixture. This is the precursor to aminorex itself, and using it in the cyanogen bromide reaction, according to Poos US patent, would provide a high yield of aminorex.
If piperonal (formed by dichromate oxidation of isosafrole in about 55-65% yield) were used in place of benzaldehyde in the above reaction, one would get as the final product the amino alcohol which could then be reacted with cyanogen bromide to form the MD analogue to aminorex.
--PK
(Chief Bee)
10-17-01 05:48
No 225558
Psychokitty: Give me all those refs you are talking about - seems highly interesting!
(Hive Bee)
10-17-01 06:41
No 225584
(Rated as: good read)
The only one I have handy right now is the synthesis of piperonal:
Synthesis of heliotropin from isosafrole by the dichromate method. Koh Kimhok and Roh Siante. Formosan Sci. 5, 36-8 (1951)(in English).--Isosafrole (50g) with a mixt. of 80 g of K2CrO7, 130 g H2SO4, 4 drops of HNO3, and 800 ml of H2O at 50 deg C yields 66.9% heliotropin (piperonal).
I have other CA reference and one US patent that basically mirror the above reaction. I'll post those along with all the other references you requested.
BTW, you've got mail.
--PK
(Distinctive Doe)
10-17-01 07:26
No 225604
Or this way.
High Selectivity in the Oxidation of Mandelic Acid Derivatives and in O-Methylation of Protocatechualdehyde: New Processes for Synthesis of Vanillin, iso-Vanillin, and Heliotropin. Bjorsvik, Hans-Rene; Liguori, Lucia; Minisci, Francesco.
Org. Process Res. Dev. (2000), 4(6), 534-543.
CODEN: OPRDFK ISSN: 1083-6160.
Abstract
New synthetic procedures for vanillin, iso-vanillin, heliotropin, and protocatechualdehyde starting from catechol are described. The utilization of statistical exptl. design and multivariate modeling and the mechanistic interpretation of the acid and base catalysis in the condensation of catechol derivs. with glyoxylic acid and in the regiocontrolled methylation of protocatechualdehyde and of the Cu(II) salt catalysis in the oxidative decarboxylation of mandelic acid derivs. have allowed the development of new highly selective processes.
Methylenation of pyrocatechols.
Bonthrone, W.; Cornforth, John W.
J. Chem. Soc. C (1969), (9), 1202-4.
Abstract
High yields in the methylenation of pyrocatechols by CH2Cl2 are obtained by using a polar aprotic solvent for reaction and maintaining low concns. of the pyrocatechol dianion.
Do Your Part To Win The War
(Distinctive Doe)
10-17-01 07:29
No 225606
(Rated as: excellent)
Check this out!!
United States Patent 4,190,583
Bauer , et al. February 26, 1980
----------------------------------------
Process for the preparation of 3,4-methylenedioxymandelic acid
The course of the reaction according to the invention is surprising, since it is known, from Gazz. Chim. Ital. 96 (4), 465 (1966), that the reaction of glyoxylic acid alkyl esters with alkyl-substituted aromatic compounds in a strongly acid medium indeed leads to mandelic acid derivatives, but with alkoxy-substituted aromatic compounds, such as veratrole, gives only diphenylacetic acid derivatives.
3,4-Methylenedioxymandelic acid is, inter alia, an important starting material for the preparation of the valuable aroma substance piperonal. Piperonal is prepared from 3,4-methylenedioxymandelic acid by oxidative decarboxylation (see Current Sci. (India) 27, 22 (1958)).
EXAMPLE 1
46 g (0.5 mol) of glyoxylic acid monohdrate, 61 g (0.5 mol) of 1,2-methylenedioxybenzene and 200 g (1.74 mols) of 85% strength phosphoric acid are brought together at 25.degree. C. and the mixture is stirred for 5 hours. The temperature thereby rises to 38.degree. C. In order to preserve the stirrability of the reaction mixture, 40 ml of water are added. After 5 hours, a further 160 ml of water are added, the mixture is further stirred for 10 minutes and the crystals obtained are filtered off. The crystals are dissolved in 650 ml of water and 100 ml of toluene at 85.degree. C. and the phases are separated. After washing the toluene phase several times with 20% strength sodium hydroxide solution and distilling off the solvent, 7.1 g of 1,2-methylenedioxybenzene are recovered. After cooling the aqueous phase to room temperature, 57.6 g of 3,4-methylenedioxymandelic acid crystallise out, and after concentrating the mother liquor a further 8.3 g of 3,4-methylenedioxymandelic acid crystallise out. The yield of 3,4-methylenedioxymandelic acid is 76.1% of theory, relative to 1,2-methylenedioxybenzene reacted. It can be increased further by working up the strongly acid filtrate of the reaction mixture. However, since the working up is very expensive, it is more advantageous to concentrate the acid filtrate and re-use it.
EXAMPLE 2
200 g of 90% strength sulphuric acid are added dropwise to a mixture of 148 g (1 mol) of 50% strength aqueous glyoxylic acid and 122 g (1 mol) of 1,2-methylenedioxybenzene at 5.degree. C. in the course of 40 minutes, whilst stirring. After stirring the mixture for a further 6 hours, 500 g of ice-water are added and the mixture is stirred for a further 10 minutes. The crystals which have precipitated are filtered off and worked up as in Example 1. 6.1 g of 1,2-methylenedioxybenzene are recovered from the toluene phase. 167.3 g of 3,4-methylenedioxymandelic acid, corresponding to a yield of 85.3% of theory, relative to 1,2-methylenedioxybenzene reacted, are obtained from the aqueous phase.
EXAMPLE 3
A mixture of 92 g (1 mol) of glyoxylic acid monohydrate and 122 g (1 mol) of methylenedioxybenzene is stirred at 80.degree. C. for 4 hours, 700 ml of water and 150 ml of toluene are then added and the phases are separated. After cooling the aqueous phase to room temperature, 44.7 g of 3,4-methylenedioxymandelic acid crystallise out, and after concentrating the aqueous phase a further 17.3 g of 3,4-methylenedioxymandelic acid crystallise out. After extracting the toluene phase by shaking with 100 ml of 20% strength sodium hydroxide solution and distilling off the toluene, 10.5 g of unreacted 1,2-methylenedioxybenzene are obtained. The yield of 3,4-methylenedioxymandelic acid is 34.6% of theory, relative to 1,2-methylenedioxybenzene reacted.
EXAMPLE 4
39 g (0.424 mol) of glyoxylic acid monohydrate are dissolved in 126 g of trifluoroacetic acid, and 51.7 g (0.424 mol) of 1,2-methylenedioxybenzene are added at 20.degree. C. The initially homogeneous mixture is stirred at 20.degree. C. for 3.5 hours. The crystals which have precipitated are then filtered off and worked up as described in Example 1. 10 g of bis-(3,4-methylenedioxyphenyl)-acetic acid are obtained from the toluene phase. 62.2 g of 3,4-methylenedioxymandelic acid are obtained from the aqueous phase. This corresponds to a yield of 75% of theory, relative to 1,2-methylenedioxybenzene employed. The yield can be increased by working up the strongly acid filtrate of the reaction mixture. However, it is more advantageous to concentrate the acid filtrate and re-use it.
If, instead of glyoxylic acid monohydrate, an equivalent amount of 50% strength aqueous glyoxylic acid is employed and the mixture is stirred for 6 hours instead of 3.5 hours, 8.58 g of bis-3,4-methylenedioxyphenylacetic acid are obtained from the toluene phase and 73.4 g of 3,4-methylenedioxymandelic acid, corresponding to a yield of 76.3% of theory, relative to 1,2-methylenedioxybenzene employed, are obtained from the aqueous phase.
Do Your Part To Win The War
(Distinctive Doe)
10-17-01 07:33
No 225608
(Rated as: good read)
United States Patent 4,165,341
Umemura , et al. August 21, 1979
----------------------------------------
Process for preparing protocatechualdehyde and its derivatives
Abstract
A process for preparing protocatechualdehyde or a 3-alkoxy-4-hydroxybenzaldehyde which comprises subjecting catechol or a 2-alkoxyphenol to reaction with glyoxylic acid in a basic aqueous medium in the presence of a catalyst containing one or more compounds selected from aluminium oxide, silicon oxide and hydrated aluminium oxide in an amount of not less than 0.01 g per 1 g of the starting catechol or 2-alkoxyphenol at a temperature of 0.degree. to 50.degree. C., and then oxidizing the thus obtained reaction mixture in a basic medium.
EXAMPLE 1
To 45 ml of a 2 N aqueous solution of sodium hydroxide were added 5.50 g of catechol, 14.25 g of a 20 wt % aqueous solution of glyoxylic acid and 2.50 g of aluminium oxide [manufactured by Kishida Chem. Co., Ltd., trade name: Kassei Alumina], and the mixture was subjected to reaction with stirring at 25.degree. C. for 24 hours.
The aluminium oxide was separated from the reaction mixture by filtration and thus a reaction liquid was obtained. The aluminium oxide separated by filtration was washed with 20 ml of a 1 N aqueous solution of sodium hydroxide and the washing was added to the reaction liquid. The reaction liquid was adjusted to pH 6 by the addition of a 12 N hydrochloric acid and then the unaltered catechol was extracted three times with 50 ml portions of diethyl ether. From the extract was recovered 1.61 g of catechol.
After nitrogen gas was blown into the aqueous layer obtained after extraction to remove the dissolved oxygen, 33 g of sodium carbonate was added thereto to adjust the pH value to 10.5. Further, 20 g of a powdery copper (II) oxide was added thereto, and the mixture was placed into an autoclave and subjected to reaction at 98.degree. C. for 50 minutes with stirring while the pressure was allowed to rise. The pH value of the reaction liquid after reaction was 10.0.
After cooling the reaction mixture, the copper oxide was separated by filtration. To the thus obtained reaction liquid was added a 12 N hydrochloric acid to adjust the pH value to 2. Organic substances in the solution were extracted six times with 150 ml portions of diethyl ether.
Protocatechualdehyde in the extract was determined by gas chromatography.
The conversion of catechol was 70.7%, the yield of protocatechualdehyde was 4.51 g and the selectivity (based on the catechol consumed in the first step; this meaning is applied similarly to the following examples) was 92.4%.
COMPARATIVE EXAMPLE 1
An experiment was run in the same manner as in Example 1 except that aluminium oxide was not used.
After the first step, 2.07 g of catechol was recovered. The conversion of catechol was 62.4%, the yield of protocatechualdehyde was 3.06 g and the selectivity was 71.1%.
Do Your Part To Win The War
(Distinctive Doe)
10-17-01 07:35
No 225611
(Rated as: good read)
United States Patent 4,183,861
Maggioni January 15, 1980
----------------------------------------
Process for preparing aromatic methylene-dioxy compounds
EXAMPLE 1: METHYLENEDIOXYBENZENE:
Methylene chloride (100 ml., 1.56 moles), tetrabutylammonium bromide (6.42 g., 0.02 moles) and water (200 ml.) are placed in an autoclave. To this mixture is added a total of 15 g. pyrocatechin (0.1362 moles) and sodium hydroxide (15.9 g., 0.3975 moles) in flake form in successive stages.
The reaction temperature is maintained at 70.degree. C. and the pressure within the autoclave rises to a maximum of 2.4 atmospheres. The reaction is completed within three hours.
After the reaction is completed, the organic phase is separated and excess methylene chloride is distilled off and recycled. Pure methylenedioxybenzene (13.8 g.; 83% yield) is obtained by distillation.
Tetrabutylammonium bromide remains as a distillation residue and this material can be recovered and reused as a catalyst to produce additional product.
By substituting pyrocatechoic acid (ie., 2,3-dihydroxybenzoic acid) for the pyrocatechin of Example 1, and otherwise following the procedure described therein, the product 1-carboxy-methylenedioxybenzene is obtained.
EXAMPLE 2: METHYLENEDIOXYBENZENE:
By following the procedure described in Example 1, but substituting hexadecyltributyl phosphonium bromide for tetrabutylammonium bromide in an otherwise analogous process, 11.65 g. of methylenedioxybenzene (70% yield) is obtained.
EXAMPLE 3: 1-METHYL-3,4-METHYLENEDIOXYBENZENE:
Methylene chloride (1.56 g., 100 ml.) and tetrabutylammonium bromide (6.42 g., 0.02 moles) are placed in an autoclave, and to this mixture is added a total of 24.8 g. of 4-methylpyrocatechin (0.2 moles) and caustic soda (24 g., 0.6 moles) in flake form, with agitation, at 80.degree. C. The reaction is completed within five hours.
After the reaction is completed, the product is recovered by following the procedure described in Example 1, that is, the organic phase is separated, excess methylene chloride is distilled off and recycled and 1-methyl-3,4-methylenedioxybenzene (19.4 g.; 71.3% yield) is obtained by distillation.
EXAMPLE 4: PIPERONAL:
Methylene chloride (100 ml., 1.56 moles), tetrabutylammonium bromide (6.42 g., 0.02 moles) and water (200 ml.) are placed in an autoclave. A total of 27.6 g. of protocatechic aldehyde (0.2 moles) and caustic soda (24 g., 0.6 moles) in water (20 ml.) are then added to the autoclave in stages at a temperature of 70.degree. C.
The pressure within the autoclave increases to a maximum of 2.4 atmospheres and the reaction is completed within four hours.
The reaction mixture is allowed to cool to ambient temperatures and the organic phase is separated. Excess methylene chloride is recovered from the organic phase by distillation and high purity piperonal (21 g., 70% yield) is isolated. The product is identified by gas chromatography and infra-red spectrum analysis by comparing against a pure sample.
Upon substituting tetramethylarsonium chloride, for the tetrabutylammonium bromide of Example 4, and otherwise following the procedure described therein, an identical piperonal product is obtained.
Do Your Part To Win The War
(Distinctive Doe)
10-17-01 07:49
No 225616
There I think I added a few weapons to the Hive war chest!!!
Here are some more.
United States Patent 3838051 OLD methylation reference
United States Patent 3726924 Methyeneation and prep of strong base(trisdimethlyaminomethane)
United States Patent 3436403 Methyleneation Reactions
United States Patent 2496803 Production of Veteraldehyde from Vanillin
Find these here http://patft.uspto.gov/netahtml/srchnum.
You need a TIFF viewer installed in your browser to look at them.
Later
Foxy
Do Your Part To Win The War
(Hive Bee)
10-19-01 05:02
No 226609
(Rated as: excellent)
Rhodium:
Here are the references you requested.
Synthesis of nitroalcohols
Can. J. Chem. Vol. 39 (1961) pp.1143-1147
Can. J. Chem. Vol. 41 (1963) pp.543-545
In. Eng. Chem., 32, 34 (1940)
Kamlet, US Patent 2,151, 517
Ann., 470, 157 (1929)
Synthesis of 3,4-methylenedioxynorephedrine
CA Vol. 45 9507d
And last but not least, the formation of the amino alcohols and the reduction strategy using Zn and H2SO4 is in the following reference . . .
Shit. The article does give information as to where it came from . . . just the page numbers (506-509).
Gee. I guess I'll just have to type up the relevant information myself.
First of all, there are three ways to go about forming the intermediate nitro alcohol; however, Kamlet's stands out as the best and easiest:
(c) Method of Kamlet (3). Benzaldehyde (106 g., 1 mole) was vigorously agitated with sodium bisulfite (110 g., 1.06 mole) in 500 ml of water until the formation of the addition compound was complete. Simultaneously, nitroethane (or nitromethane) (82.5 g., 1.10 mole) was dissolved in a solution made from sodium hydroxide (45 g., 1.125 moles) dissolved in 200 ml of water. This solution was gradually added, with agitation and at room temperature, to the addition product of benzaldehyde and sodium bisulfite. After stirring for a half hour, the mixture was allowed to stand overnight. The lower layer was discarded and the upper layer was dissolved in ether and washed with sodium bisulfite solution. The ethereal solution was dried over Drierite, and after removal of ether, distilled (bp 120-130 @ 2-4 mm). The usual conversion is 90-100g. (50-55%) and the yield, based on benzaldehyde which reacts is nearly quantitative.
Preparation of 2-amino-phenyl-1-propanol. (a) With zinc and sulfuric acid. Sulfuric acid (375 g of 30% acid) was added with stiring to a mixture of 2-nitro-1-phenyl-1-propanol (54.3 g., 0.3 mole), zinc dust (90 g., 1.37 mole of 80 mesh zinc), and 100 ml of 95% ethanol. The acid was added at such a rate that the temperature remained at 45 deg C or below. Usually 10 to 12 hours were required. Agitation was continued for 1-2 hours after completing the addition of acid, then after extracting the acidic solution with ether to remove non-basic materials, a large excess of sodium hydroxide (as a 50% solution) was added. The product which was freed was extracted with ether. Three extractions, with a total of 500 ml of ether, sufficed. The ether solution was dried, ether was removed, and the product was distilled (b.p. 122 deg C at 4 to 5 mm); 29-32 g resulted (yield 65-70%). The viscous liquid solidified on standing, and m. 46-50 deg C.
According to the article, "The unmethylated amino alcohol was obtained by reduction of the nitro alcohol either with zinc and sulfuric acid, tin and hydrochloric acid, sodium amalgam and acetic acid . . . Presumably many of the ordinary metal-acid reducing agents can be used provided the temperature is kept sufficiently low to prevent dehydration."
The above quote implies to me that aluminum amalgam might serve handsomely in the above reaction.
Oh, yes--the CA ref is as follows:
"The iso form of 3,4-methylendioxynorephedrine was obtained by the reduction of a condensate of piperonal, EtNO2, and KHCO3 (yield 45%) with Zn dust and 33% H2SO4 in 75% yield."
Since it seems that not a fucking one of you bees has Poos' original patent, here are the details to the in situ formation of cyanogen bromide:
From Example II, Line 10:
"A solution of cyanogen bromide is prepared from 1.58 g (0.036 mole of sodium cyanide and 5.7 g (0.036 mole) of bromine in methanol . . ." Then it's used as is.
Two more syntheses of piperonal are detailed in the following references:
CA 9061 between 8 and 9 (1938)
CA Vol. 51 16572d
That's all for now.
--PK
BTW, Rhodium, are you NOT getting any of my PMs to you? I have yet to hear one fucking peep from you at all. And I believe the references that I sent you are indeed valuable. It would be nice to at least know that you've gotten them safely and securely.
(Hive Bee)
10-19-01 08:09
No 226671
P/K: Don't worry, I haven't heard a peep from him either. I don't think anybody is.
(Stoni's sexual toy)
10-19-01 12:16
No 226720
You guys won't believe the amount of PMs some of the moderators receive every day.
(Chief Bee)
10-19-01 16:40
No 226762
I got 10 PM's in the last 12 hours, and I have 50-60 unanswered ones in my in-box. I got your message PK, I have just not had the time to answer it yet. The references looked good, and than you immensely for this addition you just posted. Now we can all make substituted phenylpropanolamines to use with this reaction.
(Hive Bee)
10-20-01 23:05
No 227266
Rhodium:
Thanks for taking a load off my shoulders. For awhile there, I was thinking I was the victim of some conspiracy.
Something I realized when I was away from the computer:
The first two Canadian Journal references are GOLD!!! I hadn't really looked at them in over two years and didn't really know just how valuable they are. Well, was I wrong not to include the relevant experimental details in this thread.
In both papers are given the details to synthesizing a number of substituted nitroalcohols, the 3,4-MD derivative included, in very good yield using a RIDICULOUSLY simple synthesis. I'll post all the detailed information at a later date, but for now, here is what I remember:
A quantity of a benzaldehyde is dissolved in 95% EtOH along with an excess of nitromethane (or nitroethane). To this mixture is added a dilute solution of sodium hydroxide which may be at reduced temperature, but I don't think so. The mixture is stirred (for most benzaldehydes) about sixty seconds--that's right, ABOUT SIXTY SECONDS--and then the mixture is quenched with 5% acetic acid and left to stand. The formed nitro alcohol is isolated and then recrystallized if necessary.
I'll be back soon with the reference.
--PK
(Hive Bee)
10-20-01 23:31
No 227281
(Rated as: good read)
Doooooooooooooooode!!!! This is so fucking easy!!!
Taken from Canadian Journal of Chemistry Vol. 39, 1961 pp1143-47:
Experimental:
"General Procedure
(i) Nitroalcohols.--Aqueous sodium hydroxide (10%; 1.05 mole)was added with vigorous stirring to a mixture of the aldehyde (1.0 mole) and nitromethane (2-3 mole) dissolved in 95% ethanol at ca. 5 deg C.; the reaction mixture was vigorously stirred for a short time (under 3 minutes), accurately timed on a stop watch. Aqueous acetic acid (2%) was added to arrest the reaction and decompose the sodium derivative of the nitroalcohol; the crude product precipitated out as a yellow or colorless solid or oil. After being allowed to stand at 4 deg C for 4-5 hours, the crude product was filtered and purified by repeated crystallization from a suitable solvent.
They used many substituted benzaldehydes in this reaction but no 3,4-dimethoxy or methoxy derivatives. However, 3,4-methylenedioxy-a-nitromethylbenzylal
1.) 10 g. of piperonal (piperonaldehyde)
2.) 11 ml of nitromethane
3.) 135 ml of 95% ethanol
4.) 20 ml of 10% sodium hydroxide
5.) 160 ml of 2% acetic acid
6.) Reaction time was 60 seconds
7.) Yield of purified product was 9.3 grams
8.) Product was obtained as fine colorless needles from benzene
9.) mp was 98 deg C
10.) Literature mp was 91, 95-96, 91, and 94 all taken from different sources.
Note: All benzaldehydes that had a phenolic group on the benzene ring failed to react to form the expected nitroalcohols.
Also, using mineral acids in place of acetic acid to free up the sodium derivatives of the nitroalcohols led to some dehydration of the alcohol group to form the nitroalkenes. With a few modifications, this could end up as a good way to substituted nitroalkenes.
--PK
(Chief Bee)
10-20-01 23:42
No 227284
This good way to nitroalkenes has already been published on my page: ../rhodium /mdp2np.
(Hive Bee)
10-23-01 05:22
No 227925
(Chief Bee)
10-23-01 12:58
No 228000
Not your nitroalcohol synth, but one using NaOH/HCl to give nitroalkenes. That is good, not bad.
(Ubiquitous Precursor Medal Winner)
11-02-01 12:59
No 232086
Re United States Patent 4183861
The reaction temperature is maintained at 70.degree. C. and the pressure within the autoclave rises to a maximum of 2.4 atmospheres. The reaction is completed within three hours.
That seems to bee within the stability range for the SRV. Your two-liter polycarbonate autoclave.
turning science fact into <<science fiction>>
(Hive Bee)
11-04-01 01:50
No 232790
Okay, now to clear up a few things.
Rhodium: I've been pretty confused lately because I couldn't understand why dimethylaminorex could be synthesized by BOTH the cyanate and cyanogen bromide routes. Until, of course I figured out--by carefully examining the numbering scheme used--that 3,4-dimethylaminorex is (according to J. Med. Chem. Vol. 6, May, 1963, pp.266-272) 2-Imino-3,4-dimethyl-5-phenyloxazolidine
Of this I am certain. The paper claims that using ephedrine in the standard cyanogen bromide cyclization reaction will yield an imine substance (namely this '3,4-dimethylaminorex') which does NOT belong to the aminorex family of compounds and which has the same mp as the 3,4-dimethylaminorex on you page (71-73). This is very important because such a structural change negates most of the valued pharmacological effects of the aminorex class of drugs.
Just for informations sake, at the end of the article, the authors indicate that the appetite supressant pharmacology of each aminorex analogue varies considerably. They also allude to the unique cardiovascular and CNS effects and claim that such effects would be the primary focus of an upcoming paper:
"Reference 17: J. F. Gardocki and J. Yelnosky, manuscript in preperation."
I wonder if they ever wrote the damn thing. A catalog of various aminorex analogues and thier corresponding CNS pharmacology would be most interesting and useful.
Anyway, the authors synthesized the "3,4-dimethylaminorex" by way of reacting cyanogen bromide and ephedrine. They also formed another imino compound from the same reaction using phenylephrine as the starting material. So, it is obvious that 2-alklamino aminorex compounds cannot be synthesized by reacting amino alcohols with cyanogen bromide.
However, the author indicated an alternative synthesis whereby an isocyanate was reacted with ephedrine which then formed an intermediate urea, which was then chlorinated with thionyl chloride and then cyclized by boiling in water.
The original Poos U.S. Patent 3,161,650 indicates that although the intermediate ureas were best converted into the 2-alkylamino substances via thionyl chloride (via a displacement reaction) "other reactants susceptible to the type of reaction may also be used as, for example, hydrohalic acids such as hydrochloric acid, hydrobromic acid, or hydriodic acid . . .".
Sooooooo, it would appear that if the N-carbamyl-pseudoephedrine listed on the first page of this thread is indeed the intermediate urea Poos et al is talking about, then it should--with either thionyl chloride or hydrochloric acid (which, of course they use)--react through displacement and cyclization to form the expected 2-methylamino-aminorex.
Again, however, that doesn't seem to be the case. On the first page of this thread, Rhodium, N-carbamyl-pseudoephedrine is NOT converted to 2-methylamino-aminorex like one would think but instead to 3,4-dimethylaminorex. Either you typed up the information incorrectly, or pseudoephedrine UNLIKE ephedrine forms the imino substance when used in the cyanate reaction instead of the 2-alkylamino compound.
BTW, according to the J. Med. Chem. paper potassium cyanate is effectively used to convert 2-amino-phenylethanol into the intermediate urea. If my guess is correct, this can be cyclized into aminorex, making the cyanate reaction a good way to get to aminorex itself.
In the paper, the authors list former ways utilized to get to aminorex via various starting materials:
Reacting styrene oxide with sodium cyanamide led to a mixture of products from which a low yield of aminorex was isolated: Ann. 467, 240 (1928). Perhaps using a PTC in this reaction could help to raise the yields and overall efficiency of this reaction scheme. Using stabilized cyanamide solutions and cyanamide itself under various cond
From styrene dibromide and urea: (J. Pharm. Japan 449, 561 (1919).
For those curious about the numbering scheme used to determine the structure of the heterocyclic ring: one starts at the only oxygen ether linkage; two is the next amino group at the top point of the pentagonal structure(there is a double bond between number two amino group and number three amino group); three is the next amino group; four is the first methyl group; and five is the only methyl group directly attached to the benzene ring.
So the million dollar question is this: Does N-carbamyl-pseudoephedrine cyclize with HCl to form 2-methylamino-aminorex or 3,4-dimethylaminorex?
--PK
(Hive Bee)
11-04-01 22:04
No 233040
I've made a mechanism for the condensation of urea to 1-phenyl-1-hydroxy-2-aminopropane.
First: activation of urea
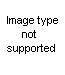
Second: attack of HNCO to norephedrine
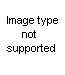
Here you are. What about it?
JustATouchOfMojoHand
(Chief Bee)
11-05-01 02:39
No 233128
Psychokitty: You are correct, the 3,4-dimethylaminorex I talk about is 4-MAR with a methyl group on the heterocyclic nitrogen (and thus the compound cannot form the two imine/amine resonance structures that aminorex and 4-MAR can do.
Do I have a 3,4-dimethylaminorex compound on my page? Where?
Correct, any 2-alkylamino aminorex compound cannot be formed either with CNBr or potassium cyanate, but it can through alkyl isocyanates, followed by cyclization of the intermediate urea (either with HX or with SOCl2).
I don't understand your confusion regarding the cyclization of N-carbamyl-ephedrine. This compound definitely cyclizes to 3,4-dimethylaminorex, and not to 2-methylamino-aminorex. Note that in the cyclization the urea nitrogen becomes the 2-amino group, and the ephedrine nitrogen ends up in the 3-position (with a methyl group attached).
Ephedrine can be converted with KOCN to 3,4-dimethylaminorex, while pseudoephedrine instead forms an amide (due to stereoconfigurational reasons), no 2-alkylamino compounds can be formed either with KOCN/CNBr.
Correct again, 2-amino-phenylethanol/KOCN should form an urea that can be cyclized into aminorex.
Could someone please find that old journal (J. Pharm Japan)? I would also appreciate any other articles regarding phenylpropanolamine derivatives which are reacted with KOCN to form ureas, followed by cyclization.
PEYOTE: What is your reference for that reaction? I did not know that urea could be transformed into HOCN like that?
Edited to fit the changed stereochemistry in the first post in this thread.
(Hive Bee)
11-05-01 21:03
No 233299
I havent got (for now) any ref for that rxn, because I've done it just this afternoon in laboratory:
I've made a phenylurea from an aniline derivative (phenetidin) with urea and HOAc,
and I can assure you that THAT'S correct (have you ever heard about synthons, or sinthetic equivalents?)
I'll serch for that refs,
but I've got a lot of things to do and very little time to do that.
Sorry for the 2nd part, I'ven't read it.... eh eh...
(Hive Bee)
11-06-01 05:56
No 233539
(Rated as: excellent)
Rhodium: Sorry for the confusion. I went back and looked at the J. Med. Chem. paper and recognized where I made my mistake. It would seem that all reactions involving ephedrine or pseudoephedrine would end up forming the 3,4-dimethylaminorex (which still really isn't from the aminorex family--at least Poos doesn't think so).
I got mixed up thinking that the authors were reacting ephedrine with potassium cyanate, when in actuality, they were reacting norephedrine with methylisocyanate to form the 2-methylamino-aminorex.
The cyanate is regarded by the paper as "classical scheme" detailed in the following reference:
J.W. Cornforth, "Heterocyclic Compounds," vol. 5, R.C. Elderfield, Ed., John Wiley and Sons, Inc. New York, NY, 1957, p.384.
As for the phenylethanolamine, it doesn't seem to have any stereochemistry (maybe just dextro and levo). So since it obviously can work in forming the intermediate urea, potassium or sodium cyanate should work at cyclizing it to aminorex.
As for phenylpropanolamine, according to the Merck index, it is norephedrine (I think). Norpseudoephedrine, however, should be easily obtained from it through reaction with base or acid. Don't know the exact ratios, but I do know there are more quantitative syntheses for pseudoephedrine from ephedrine than vice versa.
--PK
(Distinctive Doe)
11-06-01 07:49
No 233601
Aren't those naming schemes FUN to try to figure out!!
They should always draw the fucking molecule IMHO
Do Your Part To Win The War
(Hive Bee)
11-06-01 22:04
No 233865
I've calculated the yield:
From 0.28 g of phenetidine hydrochloride (p-CH3CH2OC6H4NH3+Cl-) I've obtained 0.31g
of impure p-ethoxyphenylurea (theoric 0.29 g, so the yield is over 100%: 107%), this was recrystallyzed from water,
yielding 0.26g of recrystallized product (global yield: 89.65%). An IR spectra (KBr disc) confirms that
it's the desired product. Tomorrow I'll make an 1H-NMR.
(Chief Bee)
11-06-01 23:32
No 233895
If it was recrystallized from water, perhaps your compound forms a hydrate of some sort? Now for the big question, is this reaction general for all amines, or only anilines?
If it is a general reaction, then we can make 4-MAR from phenylpropanolamine, urea, acetic acid and hydrochloric acid.
(Hive Bee)
11-07-01 20:09
No 234197
No, we used water because 4-ethoxyphenylurea isn't soluble in cold water but in hot water (this is general theory...) , I dont think that it forms any type of hydrate.
Well, I'm sure this is a general procedure, not only for aromatic amines but also for aliphatic amines. I'll search for some refs.
(Hive Bee)
11-09-01 20:59
No 234920
Here you are, from my laboratory notepad:
In a 50 mL round-bottom flask fitted with a condenser, pour 0.29g of phenetidine hydrochloride (1.67 mmol) and 2 mL of distillated water. Drop a solution of NaHCO3 10% until it reach pH 6.5 (about 4-5 drops). Then put 0.39 g of urea (6,49 mmol, 4 times of the equivalent of the phenetidine hydrochloride) and 4 drops of glacial acetic acid. Reflux for 1 hour; the reaction is monitored by TLC (eluant: CH2Cl/EtOH 19:1). The white precipitate is chilled, filtered (gooch) and dessiccated overnight on CaCl2. Yield: 0.31 (over 100%!!) So I make a recrystallization using water. Yield: 0.26 g of the recrystallized product, dulcine, or p-ethoxyphenylurea; total yield of the process: 93%. This is the reaction:
p-EtO-C6H4-NH3+Cl- + NH2CONH2 =(GAA)=> p-EtO-C6H4-NH-CO-NH2
1H-NMR analysis (with DMSO-d6;TMS 0,05%):
delta proton type signal/relative area
7.94 Ar-NH-CO-NH2 singlet/2
7.44 and 6.75 aromatic H two doublet / 2 + 2
5.18 Ar-NH-CO-NH2 singlet/1
4.02 CH3-CH2-O- quartet/2
1.38 CH-CH2-O- triplet/3
(Hive Bee)
11-29-01 11:14
No 242007
This document is archived with pictures at ../rhodium /aminoox
Extracted from Chem. Reviews, 44, 447-476 (1949)
A. Pseudoureas (2-amino-2-oxazolines)
The pseudoureas, or 2-amino-2-oxazolines (XLI), are the most widely studied
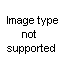
of the substituted 2-oxazolines. Equilibrium with the 2-iminooxazolidine is possible, and the chemical properties indicate many reactions in both forms. The term ‘pseudourea’ arises from the fact that these compounds are isomers of alkenylureas; thus 2-amino-2-oxazoline is isomeric with N-vinylurea and hence is ‘ethylene pseudourea’; 2-amino-5-methyl-2-oxazoline is isomeric with allylurea and is known as ‘propylene pseudourea’. The compounds are solids and strong bases, forming well-characterized salts (58). 2-phenylamizlo-2-oxazolines have been suggested for use as local anesthetics (64), but other possible applications have not developed from rather extensive theoretical studies.
1. Syntheses of pseudoureas
(a) From beta-haloalkylureas: The hydrochloride of a pseudourea is obtained upon heating the haloalkylurea with water; the addition of alkali releases the free base.
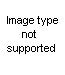
The yields vary from moderate to quantitative. The necessary unsymmetrically substituted ureas are available via several routes. One is the method of Takeda (75), who prepared pseudoureas by heating styrene dibromide, or substituted styrene dibromides, with urea. Gabriel (38) prepared unsaturated ureas from unsaturated amines and isocyanates; the addition of a halogen acid to the double bond then gave the requisite beta-haloalkylurea. The product was cyclized to the pseudourea (XLII) on heating with water and to the imidazolidone (XLIII) with alcoholic potassium hydroxide.

The haloalkylureas can be prepared by the addition of iodine isocyanate to an olefin to form a beta-iodoisocyanate, which, when allowed to react with ammonia or an amine, yields the substituted urea. This method was studied by Birckenbach and Linhard (11, 12) in the synthesis of a series of cyclohexane derivatives.

These investigators also used olefins other than cyclohexane; however, the mode of addition of iodine isocyanate to unsymmetrical olefins is not known, and the position of groups in the pseudourea is then difficult to ascertain. Thus, they were able to convert s-phenylmethylethylene to the pseudourea in 94 per cent yield, but whether it had the structure XLIV or XLV was not determined; the compound was partially resolved, the d-form being isolated.
Another route to the beta-haloalkylureas is that originally used by Gabriel (32), in which a beta-haloamine is allowed to react with cyanic acid. In this instance, the pseudourea may be obtained directly, without isolation of the intermediate beta-halourea.
4-Keto derivatives of pseudoureas are formed by cyclization of alpha-haloacyl ureas, RCHBrCONHCONHR', on treatment with alkali. Examples of such reactions have been reported by Aspelund (2a). He has also reported the conversion of 1,5-diphenyl-5-bromobarbitudc acid to a pseudourea in a reaction which apparently involves formation of an alpha-haloacylurea as intermediate. Erlenmeyer and Kleiber (26a) report the formation of 5,5-diethyl-4-imino-4-oxazolidone from guanidine and ethyl alpha-hydroxy-alpha-ethylbutyrate.
(b) From beta-hydroxyalkylureas and thioureas: The preparation of a pseudourea from a beta-hydroxyalkylurea or thiourea requires a loss of water or hydrogen sulfide in the cyclization and is otherwise similar to the preparation from beta-haloalkylureas. In using this method Soderbaum (72) cyclized a beta-hydroxyalkylurea by heating with hydrochloric acid or by heating the thio analog with alcoholic mercuric oxide. The compounds prepared were derived from s-diphenyletahanolamine through conversion to the urea or thiourea with isocyanates or isothiocyanates. The reaction was carried out where R, in XLVa, was H, CH3, C2H5, C6H5, and o-CH3C6H4.

The reaction apparently takes a deferent course when -COOR replaces -CSNHR in XLVa. Thus, ethyl N-beta-hydroxyethylcarbamate, HOCH2CH2NHCOOC2H5, loses alcohol on heating to form an oxazolidone, not a 2-ethoxy-2-oxazoline (27a).
(c) From sodium cyanamide and chlorohydrins: Synthesis of pseudoureas (see table 4) from sodium cyanamide and chlorohydrins has no similarity to the syntheses of oxazolines as do the two preceding syntheses. Fromm and coworkers (28, 29, 30, 31) have described the reaction. The mechanism is not clear, but Fromm proposes that the sodium cyanamide reacts with the water present to form sodium hydroxide, which in turn dehydrohalogenates the chlorohydrin to the epoxide. Reaction of the epoxide (XLVI) with cyanamide then follows to form an unstable intermediate (XLVII), which rearranges to the pseudourea.
This mechanism is supported by the isolation of the pseudourea from the reaction when an epoxies is substituted for the chlorohydrin. Reaction between the chlorohydrin and sodium cyanamide to give the intermediate XLVII is not excluded by this evidence. The method has been successfully used with ethylene chlorohydrin and glycerol dichlorohydrin. A yield of 33 per cent of 2-amino-2-chloromethyl-2-oxazoline was reported from the epichlorohydrin. Reaction with chloroacetic acid gave an undescribed product.
References:
(2a) Aspelund, H. : Acta Acad. Aboensis, Math. et Phys. 12, No. 5, 33 pp. (1939) (Pub. 1940); Chem. Abstracts 41, 2413 (1947); 33, 6801 (1939); 35, 2143 (1941).
(11) Birckenbach, L,.and Linhard, M. : Ber. 64B, 961-8 (1931).
(12) Birckenbach, L,.and Linhard, M. : Ber. 64B, 1076-87 (1931).
(26a) Erlenmeyer, H., and Kleiber, A.: Helv. Chim. Acta 21, 111-12 (1938).
(27a) Franchimont, .A. P. N., and Lublin, A.: Rec. trav. chim. 21, 45-55 (1902).
(28) Fromm, E., Barrenscheen, H., Frieder, J., Pirk, L., and Kapeller, R.: Ann. 442, 130-49 (1925) .
(29) Fromm, E., Kapeller-Adler, R., Friedenthal, W., Stangel, L., Edlitz, J., Braumann, E., and Nussbaum, J.: Ann. 467, 240-74 (1928).
(30) Fromm, E., Kapeller, R., Pirk, L., Hahn, A., and Leipert, T.: Ann. 447, 259-84 (1926).
(31) Fromm, E., and Honold, E.: Ber. 55, 902-11 (1922).
(32) Gabriel, S.: Ber. 22, 1139-54 (1889).
(38) Gabriel; S., and Stelzner, R.: Ber. 28, 2929-38 (1895).
(58) Menne, E.: Ber. 33, 657-65 (1900).
(64) Rose, C. L., Schonle, H. A., and Chen, K. K.: Pharm. Arch. 11, 81-9 (1940); Chem. Abstracts 35, 1522 (1941) .
(72) Soderbaum, H. G.: Ber. 28, 1897-1903 (1895).
(73) Strauss, E.: Ber. 33, 2825-30 (1900).
(74) Takeda, J.: J. Pharm. Soc. Japan 426, 691-709 (1917); Chem. Abstracts 11, 3241 (1917).
(75) Takeda, J., and Kuroda, S.: J. Pharm. Soc. Japan 449, 561-608 (1919); Chem. Abstracts 14, 179 (1920).
(Hive Bee / Eraser)
01-29-02 20:41
No 262369
I apologize for beating a dead horse.
There are several precursors mentioned: ephedrine, pseudoephedrine, norephedrine, norpseudoephedrine and phenylethanolamine. Different precursors will lead to different products.
Next, different precursors may each have varying stereoconfigurations depending on their source (OTC, synthesized, extracted, etc). Again, different starting stereoconfigurations will lead to different products.
After that, (I gather that) any of these precursors can be used in one of two routes: the cyanogen bromide route and the cyanate route. Once again, the choice of route can affect the product produced.
Finally there are several possible mentioned products: aminorex, 4-methylaminorex, 3,4-dimethylaminorex, and 4-methyl-5-phenyl-oxazolid-2-one. Each compound is of different pharmacological properties.
And, of course, the active products have different stereoconfigurations which are also crucial to their pharmacology. There is the optimal stereoconfiguration of 4-MAR, then the other configuration that's five times less potent. Then of course there is the lovely isomer of (x,y?)dimethylaminorex that causes fatal seizures.
Postscript: it is not entirely clear exactly what sources of precursors have which stereoconfigurations.
Given all these branches I am not overly ashamed that I can't get a complete grasp of the TREE, starting with PRECURSOR + SOURCE and ending with PRODUCT + PHARMACOLOGY.
Can anyone diagram or outline the "before and after" in a single post? That would bee cool
Those innocent eyes slit my soul up like a razor.
(Stranger)
04-18-02 07:30
No 298509
I was just curios since only the Levo-isomer of the amino alcohol yeilds the desired 2-amino-oxazoline, is this the same isomer which is the phenylpropanolamine of commerce?
Another thing, about the mechanism of the forementioned cyanate route.
As far as I can tell, the cyanic acid adds directly to the amine and rearranges to give the substituted urea intermediate which is isolated by recrystallization; then, the urea derivative with the alcohol grouping adjacent to it is refluxed in HCl forming the halide which then undergoes elimination of H-X by abstracting a proton from the urea group, then, this forms an Imine (and a carbocation??) and the carbcation bonds to the oxygen of the urea group forming the ring , the Imine component then abstracts a proton from the former amine component of the amino alcohol to form the oxazoline ring with the amine in the two position. Correct me if I`m wrong or just flame away.
But back to the original question about the mechanism involving stereochemistry in the cyanate route.
How does amide formation take place with the dextro isomer of the phenylpropanolamine Is this due to the relationship of the alcohol grouping to the amine?
for example with either the -,- isomer or, the
+,+ isomer the alcohol grouping is (cis so to speak) to the amino group and vice versa does this have something to do with it.
Comments and insight into these mechanisms would be greatly appreciated.
[/b] A mind is a terrible thing to educate by yourself.
(Hive Bee)
04-19-02 19:50
No 299161
Ephedrine gives the aminorex analog, while pseudoephedrine will yield the amide. So *norephedrine* would yield 4-methylaminorex while norpseudoephedrine (also known as the d-phenylpropanolamine of commerce) yields the amide.
It's the position of the OH-group that matters here.
That's why we should all try to dig up the Russian ref given by Psychokitty in the "Wanted references" thread in the novel discourse, which details the isomerization of pseudoephedrine to ephedrine.
I have the article and I'm willing to outline both reaction mechanisms.
e109
(Stranger)
04-19-02 23:08
No 299256
So when you speak of isomerization of the pseudoephedrine to the ephedrine, you are refering to the alcohol groupsin relation to the amine.
and not whether the amine is D or,L.
If this is the case thaen that can be done wih 25% hydrochloric acid.
one caveat to this procedure is that it works by converting the alcohol to the chloride and subsequent hydrolysis in a reversible reaction, which gives the isomer of the alcohol grouping.
The paper published by Emde (on rhodium`s website) is wrong with respect to the temperature at which this occurs at the temperature given (80 degrees celcius), the hydrochloric acid will dehydrate the alcohol and amine to give an aziridine which is uterly useless for our purposes.
This is evidenced by a darkening of the solution of ephedrine and hydrochloric acid.
I would say that isomerization of the alcohol grouping should be done at 40 degrees centigrate, and prolong the reaction time, also maybe play with the acid concentration a little, if that does`nt work.
(Stranger)
04-19-02 23:23
No 299263
I would be grateful if you could outline those mechanisms.
(Stranger)
04-20-02 22:44
No 299574
Another good idea which may be useful to our ends is the isolation of R,R isomer via preciitation of the sufuric acid ester.
I`ll dig up the article in my next post but essentially as rhodium stated in his first post, that either R,R or S,S isomer will work to for the aminorex via the cyanate route.
but, the R,S or S,R isomer will give an amide.
So if the phenylpropanolamine used here is racemic, then maybe this reaction to form the sulfuric ester of ephedrine
could be used in this case with phenylpropanolamine.
but in the example I will give in the next post R,S and R,R isomer of psuedoephedrine is added to chilled sulfuric acid
then this is precipitated by adding crushed ice and alcohol.
only the R,R isomer precipitated.
Which is the isomer of interest in this scheme.
Then, I suppose it could be hydrolysed by whatever means to give back the starting ephedrine, I will put this in the next post after I locate it, referenced and all.
(Hive Bee)
04-23-02 18:05
No 300459
(Rated as: excellent)
THE STEREOCHEMICAL COURSE OF THE CONVERSION OF 2-UREIDOALCOHOLS INTO OXAZOLIDINES PART I
(abstract)
N-cyano- and N-carbamyl-(+-)-ephedrine are converted quantitatively by treatment with acid into (+-)-2-imino-3,4-dimethyl-5-phenyloxazol
N-carbamyl-(+-)-pseudoephedrine on identical treatment gives (+-)-3.4-dimethyl-5-phenyloxazolid-2-one
----------------------------------------
So when you speak of isomerization of the pseudoephedrine to the ephedrine, you are refering to the alcohol groupsin relation to the amine and not whether the amine is D or,L.
If this is the case thaen that can be done wih 25% hydrochloric acid.
Well, it's worth a try on PPA. I'll get that procedure from Rhodium's. I hope the yield of this conversion is OK
I had a nice .BMP drawing that shows the difference in reaction and configuration of both the urea-compounds, but I cannot find how to import this here. Regular copying&pasting don't seem to work.
(Stranger)
04-23-02 21:55
No 300513
(Rated as: excellent)
here`s the isomerization review,from Rhodium,On heating ephedrine hydrochloride with 5% hydrochloric acid, under pressure, at 170-180°C (248) or with 25% acid, at 100°C, the compound is partially converted to pseudo-ephedrine (20, 32, 40). The conversion is reversible and an equilibrium is established. According to Emde (39), the rearrangement takes place by replacement of the hydroxyl group by chlorine, followed by hydrolysis. Oxidation of ephedrine or pseudo-ephedrine, gives benzaldehyde or benzoic acid.
Since PPA is difficult to obtain this guy had an interesting thought, on the hive here is the post,
IOC
(Stranger)
12-08-01 01:33
No 245572
Re: Akabori run Reply
An otc way to PPA, what I,ve dug up so far
Synthesis of aminoalcohols by aldol condesation of aminoacids with aromatic aldehydes.
The alanine is reduced via -COOH => -CH2-OH
Reaction between aromatic aldehydes and a-amino acids. I. New facts on the Akabori reaction. Takagi, Eiichi. J. Pharm. Soc. Japan (1951), 71 648-51. Journal written in Unavailable
abstract.
The Akabori reaction (I) (C.A. 41, 3774g) on BzH and dl-MeCH(NHMe)CO2H (II) with and without pyridine and removal of the unreacted BzH by steam distn. gave dl-ephedrine and dl-y-ephedrine. Similarly, direct heating of piperonal and II gave 2 dl-1-(3,4-methylenedioxyphenyl)-2-methyl
Fester5 sites that direct heating of 20g N-methyl-alanine
with 50g Bnz at 150deg untill fizzing stops, produces
12g of a mixture 3g ephedrine and 9g pseudoephedrine isomers.
Reactant BRN 471223 benzaldehyde
1720250 DL-alanine
Product BRN 3196917 (1RS,2RS)-2-amino-1-phenyl-propan-1-ol
-------------------------
Reaction Details
Reaction Classification Preparation
Temperature 140 řC
Other conditions Erwaermen des Reaktionsprodukts mit wss.-aethanol. HCl
Ref. 1 2262852; Journal; Takagi et al.; YKKZAJ; Yakugaku Zasshi; 73; 1953; 1086; Chem.Abstr.; 1954; 12021;
As promised, here are some more refs on the interesting condensation reaction between aromatic aldehydes and glycine/alanine:
BER 25: 3445 (1892) + 52 :1734 ('19)
ANN. 284: 36 + 307: 84
JCS 1943 ('26) + 2600 ('22)
JACS 76: 1322 ('54)
J.PHARM.SOC.JAP. 67: 218 ('47)
Most of the articles are pretty old to say the least but they contain some interesting stuff on the reaction we're interested in here. I'm especially interested in the J.Pharm.Soc.Jap article, which describes the preparation of a methylenedioxy-substituted phenylserine. But the practical way to go is definitely as mentioned in a certain patent, that is using a two-phase solvent system. This prevents the benzylidene phenylserine from crystallising and makes sure that the reaction mixture can be stirred at all times.
After decarboxylation, these phenylserine derivates turn into amino alcohols, the perfect substrates for aminoxazolines. By substituting the benzaldehyde, a lot of phenylserine and amino alcohol derivates can be made and thus a lotta aminoxazolines!
So with some 10x experiments with 20g alanine + 50g BnZ
a massive 15% return sux, any suggestions besides learn how to make nitroethane?
the sulfuric acid ester thing I can`t say will work it also is on Rhodium`s site.Esterification of ephedrine
(+)-Pseudo-ephedrin-O-sulfuric-acid-este
To 100 grams of ice-cold concentrated sulfuric acid situated in a beaker, 20 grams of natural (-)-ephedrine hydrochloride is added in about 40 portions within 10 minutes, whereupon hydrogen chloride gas is vigorously evolved. The solution is shaken under cooling with ice until all solids has dissolved. The walls of the beaker is rinsed from any residual crystals with 10 ml conc. H2SO4, the beaker is removed from the ice, and left to stand at 15 deg C for 15 minutes, whereupon the contents of the beaker is poured upon 200 grams of crushed ice in a 500 ml beaker, which also is cooled from the outside with an ice-bath. 300 ml of absolute ethanol is now added, whereby beautiful white crystal needles of (+)-Pseudoephedrine-O-sulfate ester precipitates. The crystals are filtered with suction in a small Buchner funnel, washed with a little cold water, and after air drying the yield is approximately 10g of (+)-Pseudoephedrine-O-sulfate ester.
----------------------------------------
I can`t say that will seperate the same isomer of PPA.
Beware of running temperatures too high in the isomerization scheme that temperature given is way too high!
It`s been tried before.
That akabori reaction looks shit simple!
Here`s another reference for the epimerization of ephedrines using alkali alcoholates, in this case the amino group is racemized as oppossed to the alcohol groupings being switched.
However, in
the patent literature statements have been made that the optically active
forms of the bases can be racemized by heating them with alkali
alcoholates, at temperatures ranging from 168-195°C, in the molten state,
or in a solvent
US Pat 2,152,976 - Chem Abs 33, 998 (1939)
Ger Pat 673,486 - Chem Abs 33, 4274 (1939)
Brit Pat 490,979 - Chem Abs 33, 5003 (1939)
US Pat 2,214,034 - Chem Abs 35, 754 (1941)
According to Akabori and Momotani (269), a mixture of an aromatic aldehyde
and an amino acid on heating yield alkamines. By means of this reaction,
ephedrine and norephedrine were synthesized.
J Chem Soc Japan 64, 608 (1943)
I Don`t speak japanesse but that looks really good if you don`t care about low yeilds.
But on the converse side you have to look closely at the article where they say a diferent reaction occurs with primary amino acids as opposed to the secondary amino acids it gives some sort of bis-phenyl product
A new reaction (III), differing from I, takes place on heating BzH and DL-alanine directly; PhCH2NH2, PhCH(OH)CHPhNH2 (2 dl-compds.), AcH, and CO2 are formed. It is considered that the I-type reaction occurs when the N of the amino acid is secondary and the III-type reaction when it is primary.
some research would have to be done on that.
(Hive Bee / Eraser)
05-01-02 03:27
No 303566
I had a nice .BMP drawing that shows the difference in reaction and configuration of both the urea-compounds, but I cannot find how to import this here. Regular copying & pasting don't seem to work.
you can post the image by simply uploading it to any free online storage site (UTFSE / Server Room), making the file shareable, and then posting the url along with the ubb code, eg:
please post the bmp; it would be great! and thanx for the abstract also ![]()
... always keeping in mind that time and space are relative things ...
(Hive Bee)
05-03-02 19:27
No 304612
thanks for the ref's
Yesterday i looked up the Emde article, but i was unable to get a good time:yield ratio from it, because i can understand German only poorly.
But what i did understand is that ephedrine gives on treatment with HCl chloropseudoephedrine, and this on hydrolysis gives chiefly pseudoephedrine and an amount of ephedrine .
But, if you treat ephedrine with cold, saturated (at 0śC) HCl chloro-PFED.HCl precipitates. So wouldn't it better yieldwise to isolate this first and then proceed to hydrolysis ?
I know you can separate norephedrine from PPA via its bitartrate salt, but i have to look up the details.
About the Akabori reaction, it still would be interesting if one had a large amount of each reactant (don't know about the price of alanine, though) and a good way for isolating it from the reaction mixture.
But for the moment i think i'll stay with pill extraction. Where i live, (pseudo)ephedrine is a prescription drug, PPA not.
Scarmani: I feel a bit like an idiot now but i would need a step-by-step guide for this, 'cause i'm rather internet-illiterate; don't know how to upload (only download
Hey what we need here is some sort of Hive molecule-drawing gizmo so we can all draw schemes while we post. It would be a visual enhancement and probably cause less misunderstandings.
e109
(Chief Bee)
05-03-02 22:21
No 304649
> Yesterday i looked up the Emde article, but i was
> unable to get a good time:yield ratio from it,
> because i can understand German only poorly.
Scan or type the article and send it to rhodium@ziplip.com and I'll translate it.
> I know you can separate norephedrine from PPA via its
> bitartrate salt, but i have to look up the details.
Yes, please look up this!
> (don't know about the price of alanine, though)
Alanine is so cheap it is almost free.
> Scarmani: I feel a bit like an idiot now but i would
> need a step-by-step guide for this, 'cause i'm rather
> internet-illiterate; don't know how to upload
Step by step instructions: Post 304378 (Rhodium: "Methinfo.com site as free storage for images", General Discourse)
(Chief Bee)
05-05-02 20:09
No 305169
Hey what we need here is some sort of Hive molecule-drawing gizmo so we can all draw schemes while we post.
Like this? Post 305167 (Rhodium: "Hive Molecular Structure Drawing Applet", General Discourse)
(Stranger)
05-06-02 14:40
No 305390
Ok there seems to be confusion here as to what nor-psuedoephedrine is, and what PPA is.
first of all, there are four possible stereo isomers, (R,R,,R,S,S,S,S,R), the Hcl reflux is only going to isomerize the alcohol so if it is R,S or S,R its going to change that to S,R and R,S respectively.
According to Rhodium only the ephedrine configuration will give the 2-amino oxazoline derivative via the cyanate route so by referring to the "ephedrine" configuration you are probably talking about the isomer of the amino alcohol where the amino grouping is of ths S configuration.
so isomerizing the alcohol grouping won`t do a damn bit of good!
Only by isomerizing the amino grouping by refluxing in alcohol solutions of alkali alcholates will work, to racemize the amino grouping.
I`m curious about the optical resolution of the two amino stereo isomers Rhodium alluded to earlier?
When he says they can precitated from one another by means of bitartrate salt formation/recrystallization, is he meaning Chiral tartaric acids or just plain tataric acid will work, where is the ref for that procedure ?
A digression, if alkali alcoholates are used to racemize amino groupings (which may explain the epimerization going on in the birch reduction), then can Lithium alcoholates be used as an OTC measure, or will it`s lower PKb value cause it to not do the trick.
I ask this because, Lithium is OTC, Sodium/Potassium is not.
(Chief Bee)
05-06-02 17:00
No 305423
Edited to fit the changed stereochemistry in the first post in this thread.
Norephedrine works in the cyanate route, while norpseudoephedrine does not.
Nor-pseudoephedrine has the absolute configuration R,R or S,S (OH and NH2 pointing in the same direction) depending on which optical isomer it is. Norephedrine is either R,S or S,R (OH and NH2 pointing in opposite directions).
Using molecular models as a visualisation aid, it should be obvious that isomerizing the OH group of L-norephedrine produces D-norpseudoephedrine, and D-norephedrine produces L-norpseudoephedrine.
To go from L-norephedrine to D-norephedrine you would have to change both the configuration of the OH and the NH2 group, which is not going to happen just like that.
Boiling norephedrine in HCl will thus produce a 50/50 mixture of norephedrine and norpseudoephedrine.
An interesting fact I found at http://www.pharmj.com/Editorial/20001111
In Europe, a different isomer of phenylpropanolamine known as norpseudoephedrine is used, while in the US it is ±norephedrine. In other words, they are two different drugs, he said. The difference is described in Martindale, which says that phenylpropanolamine exists in four isomeric forms: d- and l-norephedrine and d- and l-norpseudo-ephedrine. It continues: “Of the isomers, d-norpseudoephedrine is the most potent stimulant of the central nervous system and is contained in European phenylpropanolamine preparations; however, in North America only the racemic mixture of d,l-norephedrine is used. This consideration of the isomers present in a given preparation may partly explain why many of the adverse drug reactions reported in Europe describe an alteration of mental status whereas those in North America are more often compatible with hypertension.”
Articles:
A. Börner, H.-W. Krause, Tetrahedron Lett. 1989, 30, 929. A convenient synthesis of optically pure (S,S)-norpseudoephedrine
A. Börner, H.-W. Krause, J. prakt. Chem. 1990, 332, 307. (1S,2S)-2-Amino-1-aryl-propane-1,3-diols
Patents:
DD 275 675 (1988). VzH von D- bzw. L-threo-2-Amino-1(p-substituierter bzw. unsubsubstituierter phenyl)-propan-1-ole
DD 275 668 (1988). VzH von optisch reinen D- bzw. L-threo-2-Amino-1-aryl-propan-1-olen
DD 275 669 (1988). VzH optisch aktiver D- bzw. L-threo-2-Amino-1-aryl-propan-1-ole
(Hive Bee)
05-06-02 23:30
No 305532
You got me confused now too, Rhodium.
I thought the article said that N-carbamyl-ephedrine yields 4-MAR upon HCl treatment because -OH and -NHCH3 groups were pointing in opposite directions, thus the carbamylgroups are differently arranged depending on the place of -OH group.
I thought that , in the case of pseudoephedrine, ammonia was liberated and the oxazolid-2-one formed after treatment with HCl.
I'll look it all up again to be sure, we could also try it on both l-norephedrine and d-PPA if in doubt, I'll see for the bitartate separating route.
Also thanks for pointing me to the step-by-step guide I asked for and I was very happy to see that the drawing gizmo is already there !
I'll also type in the German Emde article andsend it to you around the end of the week (got a lot of work to do)
e109
(Stranger)
05-07-02 05:24
No 305640
I see, as I suspected all along that the key caveat here is the orientation of the alcohol group in relation to the amine, and not whether the amine is D or L.
Thanks for the illustration.
So if you have double racemic S,R And R,S you say that tartaric acid will form a salt and precipitate the R,R and S,S isomer leaving the others behind in solution, could you please (Master of the Bees) Post a reference for this process?
Your humble servant will be ingratiated!
I`m curios if you speak of resolution with chiral acid, or just racemic tartaric acid ?
Reference please kind sir?
Thanks.
(Chief Bee)
10-22-04 22:03
No 537262
(Rated as: excellent)
Some Extensions of von Braun (BrCN) Reaction on Organic Bases. Part I.
Salimuzzaman Siddiqui and Bina S. Siddiqui
Z. Naturforsch. 35B, 1049-1052 (1980) (../rhodium/pdf /von.braun.cn
Abstract
Some extensions of von Braun cyanogen bromide reaction have been undertaken on conessine, isoconessine and two simpler bases, dimethyl α-naphthyl amino and diethylamine. The monocyanamides of conessine and isoconessine yielded acid amides, amino-derivatives (diamines) and guanido derivatives on careful hydrolysis, reduction and treatment with ammonia, respectively. The simpler bases also formed the acid amides and diamines but failed to give the guanido derivatives under the conditions employed for conessine series. Diamines of all those bases yielded carbinol amines on reaction with nitrous acid.
____ ___ __ _
Some Extensions of von Braun (BrCN) Reaction on Organic Bases. Part II.
Malik, Abdul; Afza, Nighat; Siddiqui, Salimuzzaman
Z.Naturforsch.B Anorg.Chem.Org.Chem. 37(4), 512-518 (1982) (../rhodium/pdf /von.braun.cn
Abstract
Extensions of von Braun Cyanogen bromide reaction on Ephedra alkaloids and simpler bases have resulted in synthesis of substituted oxazolidines and a whole series of nitrogen analogues of ephedrine, desoxyephedrine and simpler amines. The general applicability and limitations of such extension of the reaction are also discussed.
The present work deals with the cyanamides of ephedrine (1), O-acetyl ephedrine (2) and desoxyephedrine (3) which were obtained as oily liquids through the action of BrCN. Attempts were made to obtain from ephedrine cyanamide an N-amide, introducing a urea moiety in the molecule, through careful partial hydrolysis with 20% hydrochloric acid. However, the resulting uniform oily product did not show the presence of amide group in the IR and NMR spectra. Ir could be identified through physical and chemical characterization, and spectral studies as 2-imino-3,4-dimethyl-5-phenyl oxazolidine (4) (yield 1 g; 100%). The protonation of the nitrile nitrogen promotes nucleophilic attack from the hydroxyl group resulting in a cyclized product. This constituted a better method of preparation than that described by Fodor et al. [G. Fodor K. Koezka, and L. Szekeres, Acta Chim. Acad. Sci. Hung. 1, 377 (1951)] which involves a rather cumbersome three steps process resulting apparently in low yield.

Reduction of 4 with zinc and hydrochloric acid furnished colourless glistening rods of 2-amino-3,4-dimethyl-5-phenyloxazolidine

In order to prevent hydroxyl induced cyclization ephedrine was converted into desoxy ephedrine by the method of Schmidt [Arch. Pharm. 251, 320 (1913)]. As a result of extension of von Braun reaction on desoxy ephedrine it has been possible to obtain from desoxy ephedrine cyanamide (3) an N-amide (8) (yield 0.98 g; 89%) through partial acid hydrolysis. On the other hand, through reduction with zinc and hydrochloric acid, a diamine (9) has been obtained (yield 0.777g; 76%). Further, the diamine has yielded a carbinol amine (10) through reaction with nitrous acid (yield 0.613g; 61%). The structure of these products as shown below, have been arrived at through spectral data and the formation of various derivatives, which are described in the experimental.
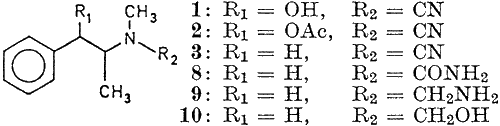
Experimental
Cyanamide derivatives of Ephedra alkaloids
The cyanamide of ephedrine and desoxyephedrine were prepared by treating their chloroform solution with freshly prepared ethereal solution of BrCN, under good cooling and mechanical stirring for 1 h. The crystalline hydrobromide formed in the reaction was filtered and washed with ether. On removal of the solvent from filtrate and washings, after washing and drying yielded cyanamide derivatives as colourless viscous liquids. The similar work up with N-methyl-O-acetyl ephedrine provided O-acetyl ephedrine cyanamide.
Substituted Oxazolidines
2-Imino-3,4-dimethyl-5-phenyloxazolidine
1 g ephedrine cyanamide was suspended in 10 ml of 20% hydrochloric acid and mechanically stirred with occasional warming till a clear solution was obtained. The reaction mixture was basified with ammonia and extracted out with ethyl acetate. The basic oily residue left on removal of the solvent was taken up in ether and treated with ethereal HCl. The resulting hydrochloride crystallized from methanol in shining needles mp 235°C.
2-amino-3,4-dimethyl-5-phenyl oxazolidine
1 g imino derivative referred to above, was heated with zinc dust and 10% aqueous HCI on water bath for half an hour the solution was filtered and basified with ammonia after prior addition of ammonium chloride. The liberated base was extracted out with ethyl acetate and after usual work up crystallized out from methanol as colourless glistening rods mp 99°C.
2-Hydroxy-3,4-dimethyl-5-phenyl oxazolidine
To a solution of 1 g amino base in 10% hydrochloric acid was added a 10% cold aqueous solution of sodium nitrite (1.2 mole) and after 15 min, the reaction mixture was extracted out with ethyl acetate. The darkish ethyl acetate layer was treated with ether and petroleum ether which threw out the resinous impurities. The nearly colourless solution gave a light yellow residue which crystallized from methanol as fluffy needles, mp 92°C.
Ephedrine-N-iminoethyl ether
1 g of ephedrine cyanamide was taken up in 10 ml absolute alcohol and to it was added 100 mg of sodium metal. After half an hour the reaction mixture was extracted out with ethyl acetate with the addition of saline. On working up the ethyl acetate extract N-imino ethyl ether derivative was obtained as colourless rectangular plates from methanol which melted at 105°C (yielded 0.782 g, 63%).
Derivatives of desoxy ephedrine
Desoxy ephedrine urea (2-methylamino-1-phenylpropane urea):
Ephedrine cyanamide (1 g) was taken in 20% aqueous hydrochloric acid and stirred at 60°C for 1 h when it went into solution. Stirring was continued for further 10 min after which the solution was cooled, basified with ammonia and extracted out with ethyl acetate. The product crystallized out from methanol as colourless shining plates mp 120°C.
N-Aminomethyl desoxyephedrine
Desoxyephedrine cyanamide (1 g) was taken in 15% aqueous HCl and heated with zinc dust on water bath till it went into solution. The heating was continued for about half an hour after which the unreacted zinc was filtered off and the filtrate ammoniated after adding ammonium chloride. The product crystallized out from methanol as colourless rectangular plates, mp 135°C.
N-Hydroxy methyl desoxyephedrine
1 g N-amino methyl derivative was taken in 10% aqueous HCl and 1.2 mole of sodium nitrite in cold aqueous solution was added on to it. The N-hydroxymethyl derivative was obtained on usual work up as colourless shining needles from methanol mp 120°C.
The Hive - Clandestine Chemists Without Borders