(Stranger)
10-12-02 02:28
No 367468
(Rated as: excellent)
Patent US6399828: Preparation of amphetamines from phenylpropanolamines.
(Chief Bee)
10-12-02 03:40
No 367477
Wow - if there ever were a "double excellent" rating, this find would be worthy of it.
(Hive Bee)
10-12-02 14:54
No 367663
It's nice to have people around here are working to advance
The cause.
(Pioneer Researcher)
10-12-02 16:18
No 367671
Very very good.
(Hive Bee)
10-12-02 17:11
No 367676
Awsome!! This opens up for some really smooth synthesis of various primary amines.
Phenylpropanolamines are readily made in high yields by CTH of 2-nitro-1-phenylpropan-1-ols, which in turn are made from the corresponding benzaldehyde and nitroethane much more easily than nitroalkenes. The O-acetyl could be inserted before the nitro group is reduced. By doing it this way the N-acetyl contaminant would not be generated.
2C-H could then be made by condensing 2,5-DMBA with nitromethane, acetylating the formed 2-nitro-1-(2,5-dimethoxyphenyl)ethan-1-o
I�ll dig up some synths of phenylnitroethanols and -propanols.
Catalytic hydrogenation freak
(Hive Bee)
10-13-02 13:23
No 367949
(Rated as: excellent)
Preparation of various phenylnitroalkanols
Patent EP0960876
Patent US2597445
Patent US1973647
Patent US1356877
Patent US4496772
Patent GB353361
Catalytic hydrogenation freak
(Pioneer Researcher)
10-14-02 17:22
No 368462
Do you think that is possible a direct CTH reduction of the acetyl nitroalcohol to the amine ? Even one pot after acetylation ?
Probably distillation will be necessary to remove the unreduced/unacetylated byproducts, but it could be a good way to make reasonable amounts with reasonable volumes.
(Hive Bee)
10-14-02 17:50
No 368471
I think it is possible. When nitroalcohol formation is complete just remove the benzaldehyde by steam distillation then acetylate. Acetylation of the isolated nitroalcohol should give 90%+ yields. The following CTH reduction of the -NO2 and hydrogenolysis of the acetoxy should give 90%+ yields as well. In the last step, just use excess of ammonium formate to push the hydrogenation/hydrogenolysis as far as possible.
Which of the useful benzaldehydes are not volatile with steam?
Catalytic hydrogenation freak
(Pioneer Researcher)
10-15-02 01:13
No 368596
If the nitroalcohol doesn't precipitate, we should wash with sodium bisulphite, but I guess it may be just filtered.
(Hive Bee)
10-15-02 09:38
No 368787
Hmmm..once again it's clear that I am not part of the Hive Illuminati.
While I can see that this process is useful, I'm having trouble figuring out exactly what the fuss is about.
For instance,
2C-H could then be made by condensing 2,5-DMBA with nitromethane, acetylating the formed 2-nitro-1-(2,5-dimethoxyphenyl)ethan-1-o
l to 1-acetoxy-2-nitro-1-(2,5-dimethoxyphenyl )ethane, which could be reduced in one step to 2C-H by CTH.
Surely it would be simpler, cheaper & OTC to synth the nitrostyrene from 2,5-DMBA & nitromethane, & then reduce with Fe/HCl ?
(particularly for those that are acetic anhydride challenged)
Mountain Boy
(Hive Bee)
10-15-02 12:18
No 368811
Mountain_Girl:
The day you get something useful from reducing 2,5-dimethoxy-beta-nitrostyrene with Fe/HCl, please let me know. The basic idea is to have as many proven methods as possible to get the goodies from as many different precursors as possible. By aiming towards this goal we will get closer to the day when we can actually hear the screams out of pure frustration from the headquarters of Mr. Hutchinfuck. Because the more methods we develop the harder it gets for them to outlaw the stuff needed to make various sorts of honey.
Catalytic hydrogenation freak
(Hive Bee)
10-15-02 13:18
No 368820
Barium:
Sorry, it was Zn not Fe that I was thinking of.
In particular :Post 217679 (sunlight: "Russian patents works!!! Zn-HCl nitrostryrene rdxn", Novel Discourse)
I agree 100% with what you said & commend everybody's efforts in this regard. It's just that not being a chemist, I sometimes struggle to understand the significance of certain posts.
Also, I felt that the above procedure appealed mainly to those who have easy access to Acetic Anhydride, Pd/C and decent apparatus, i.e. non-OTC bees.
Mountain Boy
(Chief Bee)
10-15-02 16:04
No 368850
Mountain_Girl: There is no special apparatus needed for the above procedure, as it is a catalytic transfer hydrogenation. You only need a reflux condenser and a round-bottomed flask.
(Pioneer Researcher)
10-15-02 20:28
No 368899
With the Zn HCl procedure you can easily make a 10 gr batch, and it is worth for 2CH, but is nothing if you would be playing with 3,4,5 TMNS. Thinking in a 100 gr or bigger batch, this could be interesting.
(Master Whacker)
10-16-02 02:29
No 369020
Barium,
Yes, adding the acetyl group to the nitroalcohol before reduction to the amine to prevent the formation of the amide is an excellent concept, good work! The only thing I'm concerned with in that scenario is that if acetic anhydride is employed for the esterification, you most definitely would dehydrate some if not all of the nitro- alcohol to the nitropropene. If you insist on making the acetyl ester, I recommend that the reaction be run in an ice bath using pyridine/triethylamine as solvent/halogen scavenger and acetyl chloride as the esterifying agent. A better, less watched reagent for preparation of the ester is benzoyl chloride. The benzoyl group should undergo hydrogenolysis just as in the case of the benzoylmandelonitrile to phenethylamine reduction. Other possible leaving groups are tosylates and mesylates.
(Chief Bee)
10-16-02 02:37
No 369027
Speaking of mandelonitrile esters - is there any chance they could be reduced via CTH to phenethylamines?
(Hive Bee)
10-16-02 05:05
No 369063
Would plain formic acid work in place of ammonium formate in this procedure? (The established CTH, not the theoretical 2CH preparation although that sounds particularly useful as well..) SWIM has seen several instances where formic acid is used in place of other formates; The removal of an allyl carbamate is an example, a Palladium(0) catalyst (Though usually something mroe exotic such as Palladium Tetrakis-Triphenylphosphine or Pd(dba)-CHCl3 complex) complexes with the allyl group, removing it from the protected molecule, then reduction comes into play by reducing free'd allyl group, releasing the Palladium atom. This worked best in literature with ammonium formate as the source of H- but formic acid worked with acceptable yields as well..
This could be completely un-useful for this procedure, SWIM isn't sure. :)
SpicyBrown
(Master Whacker)
10-16-02 05:10
No 369067
Unbelievably I just happened to stumble across a reference which uses acetic anhydride in a tertiary amine at 0'C to dehydrate a nitroaldol to the corresponding nitroalkene.
See: J. Org. Chem., Vol. 53, p2147, 1988 compound (8)
Now we know for a fact that acetic anhydride definitely won't acetylate the nitroaldol. Bring on the acyl chlorides!!!
Rh: I have seen a few reductions of mandelonitrile and its esters in the literature over the years. They were mostly catalytic hydrogenations and used plenty of sulfuric acid in combination with an alcoholic solvent. My thoughts on the CTH are if it can run in the presence of H2SO4 than it will probably do the trick for sure, however I have never seen a successful CTH in the lit. which used strong mineral acid as a reactant.
Perhaps mandelonitriles will reduce under standard CTH conditions and not require added acid, who'll be the first to find out?
(Hive Bee)
10-16-02 13:14
No 369209
Ritter:
Dear god I�m stupid. Of course the nitroalcohol will be dehydrated to the nitroalkene. Sigh! So it�s time to bring out those nasty acid chlorides, together with pyridine or triethylamine - the lingering stench of hardcore chemistry.
But you are, as always, right. It�s almost scary
CTH in the presence of strong mineral acids is rarely seen. This is mostly because formic or hypophosphorous acids are decomposed by HCl or H2SO4. In order to perform CTH under such conditions we need to find other donors. Cyclohexene can be used if it�s ok to work with benzene which is produced.
Spicy:
Formic acid is by far a less active hydrogen donor than its salts.
Just neutralize the formic acid with ammonium or sodium carbonate, bicarbonate or hydroxide. The potassium salts are even better hydrogen donors than the sodium salts.
Catalytic hydrogenation freak
(Pioneer Researcher)
10-16-02 20:38
No 369282
The conditions are really different if we use acetic anhydride in acetic acid or in a tertiary amine. Are you reasonabaly sure that it won't work ? Using chlorides and tertiary amines is not so attractive...
(Stranger)
10-17-02 05:54
No 369468
Acetylation of the alcohol group has been reported to work using sulfuric acid and acetic acid. This is old news. Applying the technique listed in the patent--which is diluting the acetic acid mixture with hexane; perhaps Coleman's Camper fuel might work--might be to some advantage.
But who cares?
What's more interesting is the following theoretical proposal:
1.) Apply the Friedel-Crafts acylation reaction to form the acetophenone of your choice.
2.) Brominate this using standard proceedures (1:1 aqueous methanol oxone/NaBr might work but the paper describing such a reaction seems to indicate that nuclear bromination might occur with 'electron-rich' aromatic systems).
3.) Substitute the bromine atom with hexamine (any side chain longer than that of acetophenones will not work); hydrolize with HCl to form the aminoketone-HCl. This maybe can be done in situ after the completion of the above oxone/NaBr reaction. Reduction of any excess oxidant with either bisulfite, thiosulfate, or 2-propanol, plus the addition of sodium carbonate until the solution is lightly basic would be in order to prevent hydrolysis of the hexamine into formaldehyde and ammonium chloride. This is of course assuming that hexamine is hydrolyzed by HCl only (and not by other acids such as potassium bisulfate).
Anyway, the acylophenone-hexamine complex should precipitate as such complexes are reported to be insoluble in most solvents (don't know about water though). This would necessitate that the solution be filtered and the filtered complex hydrolyzed in ethanolic-HCl to allow easy separation of the desired aminoketone-HCl. (I have a few references that detail this substitution reaction.)
4) Reduction of the carbonyl group of the aminoketone-HCl using the present CTH reaction should provide the desired phenethylamine.
Since the present patent proves that a CTH reduction of an aminoalcoholester-HCl salt can occur in water, just about any aminoketone-HCl salt should effectively be reduced to the aminoalkane-HCl salt as well. This would eliminate the need to basify the aminoketone-HCl and risk pyrazine contamination. Instead, the aminoketone-HCl from the former hexamine reaction could simply be used immediately.
What do you all think?
BTW, Rhodium. Have you checked out the only reference listed in the CTH patent? I think YOU particularly will find it most interesting as you once mentioned that you were having trouble figuring out how to synthesize a certain p-flouro compound.
(Master Whacker)
10-18-02 04:25
No 369790
Hi Regis,
That is a beautiful rxn. scheme if I have ever seen one, thank you! We need more fresh posts from people like you around here to break up the monotony of watching the same themes get rehashed time after time. Even if people don't respond to your posts publicly, TRUST ME, there are people out there who greatly appreciate and benefit from your work.
Thanks Again!
(Hive Bee)
10-18-02 15:21
No 369922
Here is another patent: Preparation of arylnitroalkanols, J Kamlet (Henry reaction is sometimes referred to as Henry-Kamlet reaction): Patent US2151517
I have an article wandering around somewhere about the synthesis of ephedrine analogues via the nitroso route. I'll try to dig it up.
EDiT
Might be helpful: V Bruckner, A Kramli; �ber eine Synthese von Ephedrinabk�mmlingen. Arch Pharm 273 (1935) 372-384. Some parts of the synthesis refer to another article by Bruckner: V Bruckner; Liebigs Ann Chem 518 (1935) in press. I don't have this one though...
EDiT 2
Stupid me... V Bruckner; �ber die Verwendung der Pseudo-nitrosite propenyl-haltiger Phenol-�ther zur Synthese von alfa-arylierten beta-hydroxylamino- un beta-amino-propanolen. Neue Beitr�ge zur Kenntnis der Acylwanderungen. Methyl-isoeugenol- und Isosafrol-Derivate. Liebigs Ann Chem 518 (1935) 226-244.
Now, that's what I call a long title
WOMAN.ZIP: Great Shareware, but be careful of viruses...
(Hive Bee)
10-20-02 13:02
No 370461
(Rated as: excellent)
Super-easy preparation
10g 2,4-dimethoxybenzaldehyde (60mmol)
5,25g nitroethane (70mmol)
30ml 90% EtOH
250mg potassium fluoride (4,3mmol)
In a 250ml rb flask with a stirbar is added 10g 2,4-dimethoxybenzaldehyde and 25ml EtOH followed by nitroethane diluted with the remaining 5ml EtOH, then finally the potassium fluoride in one portion. The suspension is allowed to stir at room temperature for 2 hours, or 30 minutes after everything has gone into solution. The solution is then decanted from some insolubles and then diluted with 100ml water. This causes a clear yellow oil to fall out. Most of the water is poured off and another 100ml water is added. Within 10 minutes the oil crystallizes to a beautiful light yellow mass.
Yield: 13,24g (54,9mmol, 91,4%) 1-(2,4-dimethoxyphenyl)-2-nitropropan-1-
While I wrote this another reaction was running in the background...here it is
1-(2,5-dimethoxyphenyl)-2-nitroethan-1-o
10g 2,5-dimethoxybenzaldehyde (60mmol)
4,25g nitromethane (70mmol)
30ml 90% EtOH
200mg KF
The reaction was performed exactly as the previous one. Within 45 minutes the reaction mixture was a dark yellow clear solution, at this point 100ml water was added which caused a dark yellow oil to fall out. Within 5 minutes this oil solidified to a dark yellow crystalline mass. The yield is not known at this point but it looks every bit as good as the previous reaction.
Catalytic hydrogenation freak
(Hive Bee)
10-20-02 19:30
No 370503
Why KF? I know lithium, sodium or potassium are used in the nitroalkanol synthesis, but is there a reason why you choose for the F- salt? Would NaF, NaCl or KCl work equally well?
WOMAN.ZIP: Great Shareware, but be careful of viruses...
(Hive Bee)
10-20-02 20:12
No 370516
The F- ion is a weak base and serves the same purpose as other bases in the Knoevenagel condensation.
Edit: Oh yeah, this wasn't actually a Knoevenagel condensation. Still, that's what the KF is used for.
(Hive Bee)
10-21-02 10:33
No 370754
Yup, the KF is the weak base needed to induce the Henry condensation of the aldehyde and nitroalkane.
Catalytic hydrogenation freak
(Hive Bee)
10-21-02 18:37
No 370871
(Rated as: excellent)
Slick �n easy
10g 1-(2,4-dimethoxyphenyl)-2-nitropropan-1-
5,2g Zn (80 mmol), activated with 5% aq HCl for 2 minutes
6,3g ammonium formate (100 mmol)
40ml 90% EtOH
To a 250ml rb flask with a magnetic stirbar containing 40 ml 90% EtOH the nitroalcohol and the activated zinc was added in one portion at room temp. Stirring was started and the ammonium formate was added in three portions over 5 minutes. The temperature rose to 55 deg C within a couple of minutes but no cooling was applied and the exotherm was allowed to run its course.
When the temperature returned to 25 deg C the almost red solution was decanted from the remaining zinc and transferred to a separatory funnel. 50ml toluene and 250ml water was added and the pH was lowered to 2 with conc HCl. The aqueous layer was extracted with another 50ml portion toluene. The bright yellow aqueous phase was then made alkaline with NaOH which caused a yellow oil to fall out. The aqueous phase was then extracted with 2x50ml toluene, the toluene extractions combined and dried over MgSO4 and the solvent removed in a rotovap. The residue is a clear yellow oil weighing 6,3g. Crystallization will be performed later.
Yield 6,3g 1-(2,4-dimethoxyphenyl)-2-aminopropan-1-
Catalytic hydrogenation freak
(Hive Bee)
10-22-02 16:07
No 371253
(Rated as: excellent)
Look what I found
Tet. Lett. No. 35, pp 3219-3222 (1978)
Nitroalkane synthesis, a convenient method for aldehyde reductive nitromethylation
The ability of fluoride ion to form very strong hydrogen bonds as HF2-, together with our expectation that the nitromethane condensation would proceed rapidly in protic solvent, led us to explore the use of potassium fluoride in IPA. In practice, we found that condensation of nitromethane with aldehyde proceeds rapidly using potassium fluoride (0.05 eq) in IPA (0.5 M), forming beta-hydroxynitroalkanes in high yield. This condensation can be further accelerated by adding a catalytic quantity (0.05 eq) of 18-crown-6.
Following the condensation, the solvent is removed leaving nitroalcohol, usually as the only detectable product. Acylation proceeds rapidly with acetic anhydride using 4-dimethylaminopyridine as a catalyst (25�C, 30 minutes). Forming of the acetates using 4-dimethylaminopyridine is superior to concentrated sulphuric acid as catalyst. Finally, reduction of acetates occurs uniformly with sodium borohydride in EtOH forming the nitroalkanes. The experimental details are illustrated for the conversion of 2-cyclohexenylacetaldehyde to 3-(2-cyclohexenyl)-1-nitropropane.
To a solution of 2 mmol aldehyde in 2 ml IPA was added 0.1 mmol potassium fluoride and 4 mmol nitromethane. After 6 h at 23�C, tlc showed one spot for the nitroalcohol at Rf 0.20 (silica gel, CHCl3). The solvent was removed at aspirator pressure and replaced with 4 ml dry Et2O. A mixture of 2.5 mmol acetic anhydride and 0.1 mmol 4-dimethylaminopyridine was added. After 8 h at 23�C formation of beta-nitroacetate was complete (Rf 0.59; silica gel, CHCl3). Finally evaporation of the ether and addition of 4 ml 1M ethanolic sodium borohydride with stirring for one hour completed the sequence. The mixture was acidified with dilute hydrochloric acid, extracted with ether, and the crude product was purified by column chromatography on silica gel to give 78% of the desired nitroalkane.
Edit: Full article in Post 442875 (Rhodium: "One-pot Reductive Nitromethylation of Aldehydes", Novel Discourse) /Rhodium
Catalytic hydrogenation freak
(Hive Bee)
10-22-02 17:54
No 371278
Patent EP0960876
Catalytic hydrogenation freak
(Chief Bee)
10-22-02 18:14
No 371284
I really like that reductive nitromethylation of aldehydes! It is a three-step one-pot procedure, something that I haven't seen much of in the literature. Do you think it would be possible to take it further than that to the phenethylamine with Red-Al or some CTH, making it a reductice aminomethylation?
(Hive Bee)
10-22-02 18:26
No 371286
If the nitro group is reduced in the presence of one eqvivalent formaldehyde - voila N-methyl whatever. Zn/HOAc is known to accomplish this. I dont know about the yields but I can look them up.
Catalytic hydrogenation freak
(Hive Bee)
10-22-02 19:45
No 371325
10g (60 mmol) 2,4-dimethoxybenzaldehyde
4.27g (70 mmol) nitromethane
200 mg KF
15ml IPA
All added to a 50ml rb flask and has been stirring for three and a half hours now. In the beginning it was a thick suspension, now it�s a clear yellow solution. I don�t have time to do a tlc right now (have to run), but from the looks of it it�s almost complete. Damn I like these small volumes of solvent
Catalytic hydrogenation freak
(Master Whacker)
10-23-02 05:44
No 371597
Barium & Crew,
Does anyone know if NaF is a strong enough base to catalyze the Henry reaction the same as KF? If not, then there's only one way to find out...."Experiment is King"
(Stranger)
10-23-02 18:30
No 371790
I looking at the article on Rhodium's page, i can only see where they cited 1g 10% pd/c. Now if the procedure is scaled up. How much 10% pd/c would be used.
(Hive Bee)
10-23-02 19:36
No 371818
Do you think we know all the articles on his site by hard?
Which procedure are talking about?
Usually it is enough with 5-10% w/w catalyst/substrate
Edit
I read the article again and realized they actually don�t mention how much catalyst they used.
Fuckers!! Well as I said earlier 5-10% w/w will be enough.
Catalytic hydrogenation freak
(Master Whacker)
10-24-02 06:33
No 372041
Barium,
Please check out: Tet. Let. 29, 5733 (1988)
This is a reference for a CTH of the nitroaldol from nitroethane and benzaldehyde producing PPA in 87% yield!!! Hydrogen donor is HCO2NH4, Pd/C in methanol. I don't have access to this article, someone PLEASE check it out!
(Hive Bee)
10-24-02 09:21
No 372070
I wanted to synthesize some aspirine the other day, using the following igredients:
- 11.5 g 3,4,5-trimethoxybenzaldehyde (M = 196.2 g/mol, purity > 98%)
- 5 mL nitroethane (M = 75.0 g/mol, d = 1.05 kg, purity > 97%)
- 30 mL EtOH (absolute)
- 168 mg NaF (M = 42 g/mol, purity > 99%)
I brought some of the 3,4,5-trimethoxybenzaldehyde in a RB flask (250 mL) and added 15 mL EtOH. I then added 5 mL nitroethane, followed by another 15 mL EtOH. I dropped the magnetic stirbar in the RB and while stirring, the NaF was added. After half an hour of stirring, it looked clear to me that the 3,4,5-trimethoxybenzaldehyde dissolved very slowly, so I heated the RB a little bit (a little bit, i.e. not refluxing). After another 2 hours, I stopped the stirring. The 3,4,5-trimethoxybenzaldehyde was dissolved completely, the reaction mixture was pale yellow in colour. I then decanted the reaction mixture in 100 mL cool demineralized water. Pricipitation commenced (PREP1). I decanted the water in another 50 mL cool demineralized water where precipitation continued (PREP2).
PREP1 was hard and yellowish white in colour. PREP2 was bright white. I dissolved some crystals in iso-octane and injected a 1 �L amount on GC/MS. The resuling chromatogram and mass spectrum are shown in the following figure.
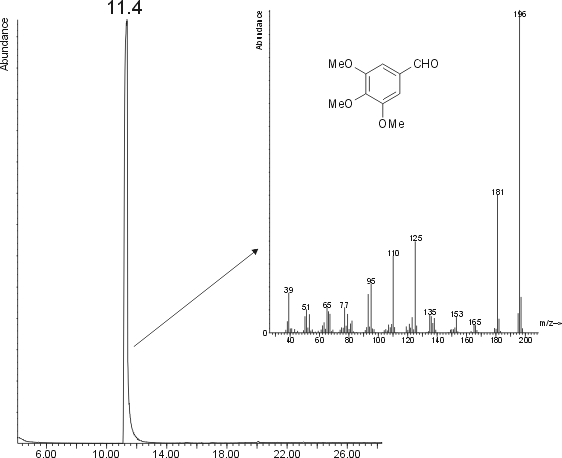
As you see, the main peak (there is only one, you cannot miss it) is a compound with M+ = 196 amu. The mass spectrum gives a 1% match for aspirine, but a 98% for 3,4,5-trimethoxybenzaldehyde
I don't know how to give these data a good interpretation... Or it is my failure (has been several months since I performed an organic synthesis), or NaF cannot be used. I'd appreciate it if another bee tries it out as well.
geh in die knie. wackle mit den hueften. klatsch in die haende. und tanz den mussolini.
(Chief Bee)
10-24-02 10:18
No 372080
3,4,5-TMNS is supposed to be neon yellow. NaF had too low catalytic activity.
(Hive Bee)
10-24-02 13:26
No 372122
He didn�t try to make 3,4,5-TMNS but 1-(3,4,5-trimethoxyphenyl)-2-nitroethan-
Catalytic hydrogenation freak
(Hive Bee)
10-24-02 14:50
No 372136
Maybe the reason can be found in Post 371253 (Barium: "Hey Ritter old chap!", Novel Discourse)?
Maybe KF in solution forms KHF2, which sets free HF2-, while NaF does not?
geh in die knie. wackle mit den hueften. klatsch in die haende. und tanz den mussolini.
(Hive Bee)
10-24-02 16:36
No 372159
That can very well be the reason why it didn�t work. Synthesis, 169 (1983) Alkali metal fluorides in organic synthesis, might shed some light over this.
Catalytic hydrogenation freak
(Hive Bee)
10-24-02 17:35
No 372175
Seems to be related to that indeed, Barium. From the Synthesis article:
"The activity of alkali metal fluorides was found to increase in the order:
CsF >= RbF > KF >> NaF > LiF
Tetraalkylammonium fluorides are also very strong bases."
"Having compared the activity of the fluorides of various alkali metals in the Knoevenagel reaction, Rand and coworkers [54] showed rubidium and caesium fluorides to be more active than potassium fluoride."
M Yield (%)
---------------
Li 0
Na ~5
K 52
Rb 77
Cs 71
Thanks Ba! Now we have something that tells us that NaF/LiF-dreams can be stocked for a while...
geh in die knie. wackle mit den hueften. klatsch in die haende. und tanz den mussolini.
(Hive Bee)
10-25-02 17:38
No 372564
(Rated as: excellent)
Tet. Lett., Vol. 29, No. 45, pp 5733-5734, 1988
Transfer Hydrogenation: A sterospecific method for the conversion of nitroalkanes into amines
Recently we needed a mild, rapid procedure for the conversion of beta-nitroalcohols into the corresponding hydroxy amines with the retention of configuration....blah blah blah...As such we consider that it is the method of choice for the production of amines from functionalized nitro alkanes.
Ba�s voice: Eight diffrent nitro alcohols were converted to the amino alcohols and the yields were around 70-85%
A typical procedure is as follows: to a solution of the nitro alcohol 1.1 mmol in 10 ml 50/50 THF-MeOH was added 50 mg 10% Pd/C, followed by 5 equiv ammonium formate. The mixture was stirred at room temperature until all nitro alcohol had been consumed (tlc). The mixture was diluted with 100ml Et2O, filtered and the filtrate was evaporated in vacuo to yield the crude amine. Flash column chromatography (SiO2, MeOH/CHCl3 2:98 v/v) gave the amine.
Catalytic hydrogenation freak
(Hive Bee)
10-25-02 17:48
No 372570
I�ve tried the above method with 2-nitro-1-(2,4-dimethoxyphenyl)-1-ethano
But I do by no means give up this easily. Tomorrow Ra-nickel will be used instead.
Catalytic hydrogenation freak
(Hive Bee)
10-26-02 15:19
No 372851
10g (60 mmol) 2,4-dimethoxybenzaldehyde
4.27g (70 mmol) nitromethane
15ml IPA
No KF added
All was added to a 100ml rb flask and stirred for 40 hours at room temperature. Some of the benzaldehyde had gone into solution. The solution was decanted from the undissolved material and diluted with 100 ml water. This caused a yellow oil to fall out which solidified within seconds. The mp was 62-64�C (mp. pure material 69-72�C), probably depressed due to the presence of nitromethane and IPA.
Catalytic hydrogenation freak
(Hive Bee)
10-26-02 17:39
No 372879
These patents cover the process of making nitroalkanes, the same way as in the article above, and nitroalcohols using different catalysts, trialkylphosphines.
Patent US3723546 - nitroalkanes
Patent US4496772 - trialkylphosphine
Edit
Reduction of isonitrosopropiophenones with H2/Pt/Pd
Patent US3028429
Catalytic hydrogenation freak
(Hive Bee)
10-29-02 13:35
No 374027
(Rated as: excellent)
The following procedure contains information on the formation of 3,4,5-trimethoxyphenyl-2-nitropropanol from nitroethane and the 3,4,5-trimethoxybenzaldehyde bisulfite adduct. It should be used for information services only. Every clandestine abuse is disencouraged.
Used products:
- 5 g 3,4,5-trimethoxybenzaldehyde (M = 196.20 g/mol, n = 25.5 mmol)
- 2.5 mL nitroethane (M = 75.07 g/mol, d = 1.05 kg, n = 35 mmol)
- 7 mL NaHSO3 (39%)
- 1.1 g NaOH in 10 mL H2O
Working procedure:
A 100 mL RB flask is charged with 5 g 3,4,5-trimethoxybenzaldehyde and 10 mL water. Stirring is commenced and 7 mL bisulfite is added. The adduct forms slowly (since the benzaldehyde flakes won't dissolve very well), so the reaction mixture is heated until everything has gone into solution. At this point, the reaction mixture is somewhat dark yellow in colour. The heating is stopped and 2.5 mL nitroethane is added, immediately followed by 1.1 g NaOH in 10 mL water. The mixture was stirred overnight. When the reaction was halted, a yellow oil settled at the bottom of the RB flask. The oil was isolated and added to a small amount of NaHSO3 solution to recover some trace amounts of 3,4,5-trimethoxybenzaldehyde (and to purify the oil at the same time). The mixture was stirred for 20 minutes, after which the yellow oil was isolated again. It was dumped in 75 mL H2O and put in the fridge (at 4 C). When I checked after a couple of hours, a yellow crystalline cake had formed. After drying, the weight of the cake was 6.1 g, or a yield of 88%.
Remarks:
(1) Using the bisulfite adduct means we don't have to worry about the benzaldehyde auto-oxydation cascade reaction, or in simpler words: no inert atmosphere required.
(2) As you might have read, I purify the oil with bisulfite solution. The reason for this is bivalent: as said, the oil is purified from benzaldehyde remainings (thus you obtain a very pure product without recrystallization), but more important is that you can recycle unreacted benzaldehyde traces for a next run. This means that the 88% yield from the first run is only increased if you use the recycled benzaldehyde in a second run.
(3) I think this reaction is easily upscaled...
geh in die knie. wackle mit den hueften. klatsch in die haende. und tanz den mussolini.
(Hive Bee)
10-29-02 16:45
No 374076
This is the old method for the condensation of nitroalkanes with benzaldehydes. It should be no problem to scale it up to huge quantities. I remember from one of the patents I posted earlier in this thread that they gave an example of a batch yielding 17 kg 2-nitro-1-phenylpropan1-ol. But I would be a little concerned about handling such huge amounts of the sodium or potassium salt of nitroethane. Even though it is in a aqueous solution.
The removal of the benzylic -OH group by forming an ester doesn�t necessarily call for acetic anhydride. Propionic or butyric anhydride should work just as well. I don�t believe any of them are regulated. Or are they?
Catalytic hydrogenation freak
(Hive Bee)
10-29-02 17:36
No 374083
Kamlet's patent uses 10.7 kg (!) of benzaldehyde, that's why SWiM "hinted" it
Currently testing:
- benzaldehyde
- anisaldehyde
- 3-methoxy-4-hydroxybenzaldehyde
Results will follow...
geh in die knie. wackle mit den hueften. klatsch in die haende. und tanz den mussolini.
(Hive Bee)
11-18-02 00:25
No 380564
I just found something on the net which I had to share...
Water-Soluble Nickel Catalysts:
From 2-Pyridylphosphine to �Ligand Free� Species
Matthew D. Le Page and Brian R. James*
Department of Chemistry, University of British Columbia, Vancouver,
British Columbia, V6T 1Z1, Canada
Interest remains high in development of homogeneous catalysis systems that are effective in
aqueous solution. Contributions from this laboratory have focussed on the use of the 2-
pyridylphosphine ligands PPh3-xpyx (or PNx, where x is the number of 2-pyridyl groups),
py2P(CH2)2Ppy2 (1, the analogue of diphos), and py2P(C5H8)Ppy2 (2, where C5H8 is a
cyclopentane backbone).1 Diamagnetic nickel complexes synthesized include the purely Pcoordinated
species: NiX2(P-P), Ni(CO)2(P-P), Ni(CO)2(PNx)2, Ni(PNx)4, Ni(P-P)2, and
Ni(P-P)2(PNx)2, where X = a monoanion, and P-P = 1 or 2. Isolated, paramagnetic
complexes include the P-coordinated NiX2(PNx)2, the P,N-coordinated NiCl2(PN3), and the
N,N�,N�-coordinated [Ni(PN3)2]Cl2. In aqueous media, species such as Ni(H2O)2(P-P)2+,
Ni(H2O)2(PNx)2
2+, and Ni(CO)(2H-P-P)2+ are formed, where 2H-P-P means diprotonation at
the N-atoms.
Representative complexes from those listed above have been tested in aqueous solution for
catalytic activity toward olefin hydration and the Water-Gas Shift Reaction (WGSR). The
only notable activity was that of NiCl2(PN2)2 for the WGSR, in which a turn-over frequency
for H2/CO2 production was ~30 h-1 at 100oC and 40 atm CO, in the range commonly
observed for homogeneous WGSR catalysts, including that noted for NiCl2(PMe3)2 in basic,
aqueous EtOH.2
During these studies, a report appeared on the use of a NiCl2(PPh3)2/NaOH/iPrOH system
for transfer hydrogenation of ketones and aldehydes.3 Our water-soluble Ni(II)
pyridylphosphines were of comparable activity, but some "blank tests" soon revealed that
the simple salts NiBr2 and NiI2 had much higher activity!
For example, 90% conversion of
cyclohexanone to cyclohexanol was attained after 1 h of refluxing in an alkaline iPrOH
solution containing NiBr2, whereas conversions of only 14 and 24% respectively, were
achieved with NiCl2(PPh3)2 and NiBr2(PPh3)2, respectively, under comparable conditions;
optimization of reaction conditions (~1.5 M substrate, 5 mM NiBr2, 0.5 M NaOH) resulted
in 100% conversion after 30 min refluxing. The simplicity of this system, which uses a
commercially available, inexpensive Ni(II) salt, makes it attractive for laboratory
hydrogenations without the need for H2.4 Of note, NiBr2(PPh3)2 in the alkaline medium
dissociates the PPh3 ligands, implying that in the reported work with the NiCl2(PPh3)2
system, the precursor catalyst may be simply NiCl2.
Studies with other acceptor substrates show that the NiBr2/NaOH/iPrOH system is effective
for catalytic transfer hydrogenation of other saturated ketones (97-100% in 24 - 48 h),
alkenes (e.g. n-octene to octane, 99% in 30 min), alpha, beta-unsaturated ketones (e.g. 2-
cyclohexen-1-one to cyclohexanol (71%) and cyclohexanone (2%) over 48 h), nitrobenzene
to exclusively aniline (19%, 24 h), and 4-nitro-benzaldehyde to a mixture of reduced
products (38%, 24 h).
[NiBr2, NaOH]
Acceptor (A) + Me2CH(OH)----->A(H)2 + Me2CO
reflux
No hydrogenation was observed for internal olefins such as trans-2-octene, and cyclooctene.
Kinetic studies on the hydrogenation of cyclohexanone, which appears to be a homogeneous
catalyzed system as judged by a mercury-addition test, reveal the following kinetic
dependences: 1st- to fractional-order on [NiBr2] with increasing concentration, 2nd order on
[NaOH], 1st-order on ketone (up to ~1.5 M), and 1st-order on iPrOH (up to ~2.0 M). Of
several plausible mechanisms, one involving hydride transfer from coordinated ispropoxide
to coordinated cyclohexanone, with a subsequent protonation to release the cyclohexanol,
satisfies the kinetics if the coordinated isopropoxide is associated within a cluster form of a
�Ni(OiPr)2� monomer. Spectroscopic data are being sought to support such a mechanism.
We thank NSERC of Canada for financial support.
References
1. N. Jones, K. S. MacFarlane, M. B. Smith, R. P. Schutte, S. J. Rettig and B. R. James,
Inorg.
Chem., 38 (1999) 3956.
2. P. Giannoccaro, E. Pannacciulli and G. Vasapollo, Inorg. Chim. Acta, 96 (1985) 179; R.
M.
Laine and E. J. Crawford, J. Mol. Catal., 44 (1988) 357.
3. S. Iyer and J. P. Varghese, J. Chem. Soc., Chem. Commun., (1995) 465.
4. M. D. Le Page and B. R. James, J. Chem. Soc., Chem. Commun., (2000) 1647.
Quidquid agis, prudenter agas et respice finem!
(Chief Bee)
11-19-02 04:11
No 381011
J. Chem. Soc. Chem. Commun. 1647-1648 (2000) (../rhodium/pdf /cth.nickelbr
(Hive Bee)
11-19-02 17:36
No 381224
Thanks for the input, Rhodium. I wonder about if this reaction could be employed for the reduction of our beloved nitropropenes. The C=C bond should be reduced, but what about the (aliphatic) nitro group? Yields of amines from aromatic nitro compounds (e.g. anilines) was really low and aliphatic nitro groups are somewhat harder to reduce to the amino group. Any comments?
Quidquid agis, prudenter agas et respice finem!
(Hive Bee)
01-04-03 18:07
No 395403
Currently a 300 mmol batch of 2,5-dimethoxybenzaldehyde --> 2C-H is being tested. The final reduction will be done tomorrow but here is what�s happened so far:
50 g 2,5-dimethoxybenzaldehyde (300 mmol)
21 g nitromethane (roughly 350 mmol)
700 mg KF
75 ml 99% EtOH
All was added to a rb flask containing a magnetic stirbar and placed in a cold (12�C) water bath. Stirring was started and the reaction was left overnight. In the morning the solution was thick from a yellow crystalline mass. The alcohol and residual nitromethane was removed by distillation under reduced pressure with the water bath only heated to 42�C. The reason to be a bit conservative with the temperature is to prevent the beta-nitroalcohol to be dehydrated to the nitrostyrene. The last of the alcohol was removed by co-destillation with toluene.
When no more ethanol came over the toluene suspension (100 ml toluene was added to aid the removal of the ethanol and approximately 80 ml remained) was chilled to 15�C and 1,5 g 4-(N,N-dimethylamino)pyridine (roughly 5 mol%) was added in one portion followed by 40 ml acetic anhydride (roughly 400 mmol) in 5 ml portions. The temperature rose a bit after each addition. When all acetic anhydride had been added all of the crystalline beta-nitroalcohol had gone into solution.
So far so good, the reductions will be performed tomorrow.
Catalytic hydrogenation freak
(Hive Bee)
01-05-03 17:17
No 395584
Continued
When checking the contents of the reaction flask this morning I was quite surprised to find a quite good yield of 2,5-dimethoxy-beta-nitrostyrene instead of 1-acetoxy-1-(2,5-dimethoxyphenyl)-2-nitr
So this was not the way to acylate the beta-nitroalcohol. But let�s try the same method with one more carbon on the aliphatic chain. It�s so typical, when I don�t want the nitrostyrene I end up with a good yield of it.
Catalytic hydrogenation freak
(Stoni's sexual toy)
01-05-03 17:34
No 395585
I feel so sorry for you. 92% yield of a completely unusable precursor... I bet that sucks big time!
I'm not fat just horizontally disproportionate.
(Hive Bee)
01-05-03 18:03
No 395589
I�m gonna cry a bit tonight
Catalytic hydrogenation freak
(Hive Bee)
01-08-03 17:04
No 396415
20 g 1-(2,5-dimethoxyphenyl)-2-nitroethanol (88 mmol)
40 ml EtOH
2-3 g Raney nickel
42 g sodium hypophosphite hydrate (395 mmol), dissolved in about 40 ml water
The catalyst was added to a 250 ml E-beaker containing a magnetic stirbar and 40 ml EtOH followed by the powdered substrate in one portion. Stirring was started and 5 ml of the hypophosphite solution was added. At this point the temperature of the suspension was 16�C. Nothing at all happened. No gas evolution or increased temperature. After 5 minutes of waiting another 0,5 g Raney nickel was added. Immediately gas evolution started followed by a slow climb in temperature. When the gas evolution slowed down the temperature had climbed to 27�C. The remaining hypophosphite solution was added in portions during one hour, while the temperature increased to a maximum of 62�C. No external cooling was applied. Just before the last portion of the hypophosphite solution was added the temperature has already begun to decrease.
The reaction suspension was cooled to room temperature and the catalyst was removed by filtration through celite. The filtrate quickly seprated into two phases, a bottom aqueous solution and a bright yellow alcohol solution of the amine. The phases was separated and the organic phase was stripped of solvent under reduced pressure leaving a clear yellow oil. The bright yellow oil was dissolved in 100 ml EtOAc and about 50 ml solvent distilled off under slightly reduced pressure. To the remaining solution an additional 50 ml EtOAc was added followed by slow addition of dry HCl in IPA until crystallisation ceased. The crystals was removed by filtration and dried to constant weight.
Yield: 16 g 2-amino-1-(2,5-dimethoxyphenyl)ethanol hydrochloride (68 mmol, 77%)
Next step is to acylate this beta-aminoalcohol like the patent describes.
Catalytic hydrogenation freak
(Hive Addict)
01-09-03 06:50
No 396586
In GAA with perchloric acid as promotor and Pd/BaSO4 as catalyst? This would eliminate the need of acetic anhydride. (not important to Barium probably but for others maybe)
or do I overlook problems with the methoxy groups?
ORG
(Hive Bee)
01-09-03 14:09
No 396660
2-amino-1-(2,4-dimethoxyphenyl)ethanol hydrochloride, 62% yield
2-amino-1-(3,4-dimethoxyphenyl)propanol hydrochloride, 59% yield
2-amino-1-(3,4,5-trimethoxyphenyl)ethano
2-amino-1-(3,4,5-trimethoxyphenyl)propan
None of the intermediate beta-nitroalcohols were isolated. The alcoholic solutions of the nitroalcohols were just diluted with more EtOH and the reduction started right away. I�m sure low-pressure catalytic hydrogenations of the nitroalcohols with raney nickel in MeOH or EtOH will give higher yields.
Catalytic hydrogenation freak
(Hive Bee)
01-11-03 13:42
No 397220
(Rated as: excellent)
2-Nitro-1-(2,5-dimethoxyphenyl)ethanol
2,5-Dimethoxybenzaldehyde, 8,3 g (50 mmol)
Nitromethane, 3 ml (55 mmol)
Sodium hydroxide, 0,2 g (5 mmol)
Aliquat 336, 1,3 g (4 mmol)
Water, 100 ml
MeOH, 15 ml
The sodium hydroxide was dissolved in 100 ml water in a 250 ml beaker containing a magnetic stirbar followed by nitromethane, 2,5-dimethoxybenzaldehyde, and methanol. The finely powdered benzaldehyde started to form big lumps in the stirred solution. The PTC was added and stirring continued. Within one minute a clear yellow oil separated from the lumpy benzaldehyde. After about ten minutes of stirring the lumpy benzaldehyde was gone and the thick yellow oil covered the bottom of the beaker. As the stirring continued the oil solidified to a creamy-yellow solid over 5-10 minutes. The alkaline aqueous solution was removed by decantation, the solid washed with water until neutral, recrystallized from MeOH and dried to constant weight.
Yield: 9,1 g (40 mmol, 80%)
This was a test of the procedure found in J. O. C., vol 62, 425-427 (1997) which can be downloaded from a link on Rhodiums website. Thank you so much for the link.
Currently another trial reaction is running with 50 mmol 2,5-dimethoxybenzaldehyde and 55 mmol nitroethane.This time with no MeOH added. Damn it, this reaction looks every bit as good as the previous one.
Catalytic hydrogenation freak
(Hive Bee)
01-23-03 21:17
No 400902
(Rated as: excellent)
This might be something interesting for you Ba:
Facile synthesis of 2-nitroalkanols by tetramethylguanidine (TMG)-catalyzed addition of primary nitroalkanes to aldehydes and alicyclic ketones
Daniele Simoni et al. Tetrahedron Lett 38(15) (1997) 2749-2752
Abstract - Tetramethylguanidine-catalyzed addition of primary nitroalkanes to aldehydes and alicyclic ketones constitutes a practical means to perform the nitro-aldol reaction (Henry reaction). The very mild conditions employed, together with the short reaction times, make the procedure tolerant of a range of functionalities and highly versatile for the synthesis of a variety of 2-nitroalkanols.
Nitroalkanes are versatile building blocks and intermediates in organic synthesis, primarly due to the ease of carbon-carbon bond forming reactions of derived species such as nitronate anions, silyl nitronates, and nitrile oxides (1-4). Among nitroalkanes, 2-nitroalkanols are particularly versatile intermediates for the synthesis of nitroalkenes, 2-aminoalcohols and alfa-nitroketones; (1-3) moreover, they are of importance because of their biological activity as fungicides. (5)
Classical preparation of 2-nitroalkanols involves base-catalyzed addition of a nitroalkane to a carbonyl compound (Henry reaction), the most widely used condensing agents being alkali and alkaline alkoxides in alcoholic solution. (2,6-9) Improved procedures have been recently introduced by Rosini et al. (9-10) who effected the addition of nitroalkanes to aliphatic aldehydes on an alumina surface, and by Wollenberg et al. (11) through a fluoride ion-catalyzed reaction of nitromethane to aliphatic aldehydes. However, these methods, although efficient, are not of general applicability, being generally limited to aliphatic aldehydes. Therefore, the development of new methodologies for the preparation of 2-nitroalkanols is very attractive owing to their synthetic value.
In this letter we report our preliminary results concerning the tetramethylguanidine(TMG)-catalyzed addition of nitroalkanes to aldehydes and ketones as a practical means to perform the nitro-aldol addition reaction. Our own previous observation of the ability of TMG to catalyze the Michael addition of nitromethane to alfa,beta-unsaturated carboxylic acid esters as well as alfa,beta-unsaturated ketones (12,13) [Scheme 1] suggested to us to extend its utility to bring about the nitroaldol addition reaction [Scheme 2].
[Scheme 1]
R1
R2--C==C--C==O TMG |
| | + MeNO2 ----------> O2N--CH2--C--CH2--C==O
R1 R3 room temp | |
R2 R3
Hence, when an aldehyde (1 mmol) was solubilized at O C in nitromethane (10 mL) in the presence of a catlytic amount of TMG (two drops), the addition reaction took place smoothly giving rise to the 2-nitroalcohols in good yield (Table).(14) The reaction can be applied both to aromatic and aliphatic aldehydes simply through a slight modification of the reaction conditions (see times and temperatures in Table). Attempts to perform the reaction in organic solvents (i.e. dichloromethane, acetonitrile or toluene) in the presence of one equivalent of nitromethane, resulted in somewhat lower yields, the formation of the 1,3-dinitro derivative being concomitant. Moreover, when nitroethane was used, the nitro-aldol addition ocurred in good yields but without diastereoselectivity.
[Scheme 2]
NO2 R2
TMG | |
R1--CH2--NO2 + R2--C==O ------------------> R1--CH--C--R3
| room temperature |
R3 or O C OH
[4] [5] [6]
4a: R1=H
4b: R1=Me
On the other hand, the condensation of aliphatic as well as alicyclic ketones with primary nitroalkanes usually takes place in the presence of alkaline alkoxides in alcoholic solution. The use of aliphatic amines as catalysts has also been tried, resulting in the prevalent formation of 1,3-dinitro-paraffins or the nitroalkene derivatives (15). Furthermore, Lambert and Lowe (15) claimed the preparation of 2-nitroalkanols using triethylamine as the catalyst, without reporting any experimental detail. We observed that TMG catalyzes the Henry addition of nitromethane to cycloalkanones at room temperature producing good yield of the desired 2-nitroalkanols after the required time (see Table). Acetophenone failed to give condensation products, at least under these conditions, whereas the reaction of aliphatic ketones such as acetone, 2-butanone or 2-hexanone, resulted in somewhat lower yield possibly due to the preferential self-condensation reaction.
In summary, as shown in Table, the present methodology is particularly suitable using aldehydes and alicyclic ketones as substrates allowing an easy isolation of the desired adducts in excellent to good yields. Aromatic aldehydes gave the corresponding nitroalcohol derivatives as the only detectable products in good yields.
In conclusion, this method offers significant advantages over existing methods, especially in terms of milder reaction conditions, shorter reaction times, and that anhydrous solvents or reagents and inert atmosphere conditions are not required. Interestingly, the TMG-catalyzed addition of nitromethane to cyclohexanone afforded the corresponding nitroalchol in 71% yield, thus comparing very favourably with the methodology described in "Organic Syntheses".(16)
Finally, considering the ease of operation and the simplicity of workup of the developed methodology one may expect its widespread application for industrial purposes such as generating combinatorial 2-nitroalkanols libraries.
[Table] (note: only 'interesting' compounds shown)
1. Benzaldehyde + MeNO2 -> 94%, 30 min, 0 C
3. p-nitrobenzaldehyde + MeNO2 -> 97%, 15 min, O C
4. anisaldehyde + MeNO2 -> 73%, 60 min, O C
8. p-nitrobenzaldehyde + EtNO2 -> 88%, 15 min, O C (*)
9. anisaldehyde + EtNO2 -> 74%, 60 min, O C (*)
(*) no diastereoselectivity was observed.
Experimental procedure: a solution of 0.01 mol of the carbonyl compound in 10 mL of the primary nitroalkane was cooled at 0 C and then two drops of TMG were added. The reaction was allowed to stand at 0 C (or at r.t. in case of ketones) for the time indicated in the table, then it was diluted with brine, acidified with 5% HCl solution and extracted with EtOAc. The combined extracts were dried over anhydrous MgSO4 and after removing the solvent the pure 2-nitroalkanol was obtained by distillation or by flash chromatography.
- The END -
Ave Hive, synthetisandi te salutant!
(Hive Addict)
01-24-03 16:25
No 401121
(Rated as: excellent)
Thank you GC_MS. May I tempt you with this one;
Tetrahedron, 52, 1677-1684 (1996)
Nitroaldol (Henry) reaction catalyzed by Amberlyst A-21 as a far superior heterogenous catalyst
...We found that Amberlyst A-21 is far superior as general catalyst for the nitroaldol reaction [1], in fact it is possible to obtain beta-nitroalkanols in high yields (70-95%), by limited reaction times, and from a wide variety of starting materials. The Amberlyst A-21 avoids the dehydration of the 2-nitroalcohols into nitroalkenes even if aromatic aldehydes are used.
The reaction has been performed by adding 8-10 g of Amberlyst A-21 to 50 mmol of aldehyde and 50 mmol of nitroalkane, however, unlkie other methods the yields are substantially independent from the ratio catalyst/starting materials. It is important to point out that, after recycling, the catalyst can be reused withouta considerable loss of efficiency. Different solvents can be used (Et2O, CH2Cl2, THF) without a substantial change of the yield.
By our method both primary and secondary nitroalkanes gives god results. Compared to other processes our procedure gives, generally, better yields. Moreover, this catalyst does not affect labile funtional groups and its mildness is demonstrated by the stablilty of the bromohydrin to epoxide formation, under basic conditions.
General procedure for the synthesis of nitroalcohols, without solvent.
A 100 ml two-necked flask equipped with a mechanical stirrer was charged with the nitro compound (30 mmol) and cooled with an ice-water bath. The aldehyde (30 mmol) was added, and the mixture was stirred for 15 minutes. Amberlyst A-21 (5-7 g) was added, and stirring cotinued for the right time. The Amberlyst was washed with CH2Cl2 (4 x 25 ml). The filtered extract was evaporated and the crude nitroalcohol was purified by chromatography or used as it is.
General procedure for the synthesis of nitroalcohols, with solvent.
The nitro compound (30 mmol) and the aldehyde (30 mmol) were added to the solvent (30 ml), then Amberlyst A-21 (5-7 g) was added and the mixture was magnetically stirred for the right time. After filtration the Amberlyst was washed with the solvent used and the extracts were evaporated. The crude nitroalcohol was purified as above.
[1] From J. Chem. Soc., Perkin Trans. 1, 107-110 (1999)
...The utilisation of Amberlyst A-21 proved to be not very efficient in our hands. Instead of Amberlyst A-21, Amberlite IRA-420 (OH-form) or DOWEX-1 (OH-form) can be used.
Barium�s voice In this article the authors first converted benzyl alcohol, p-MeO-benzyl alcohol, p-NO2-benzyl alcohol, p-F-benzyl alcohol and o-F-benzyl alcohol to the benzaldehydes by oxidation using polymer supported permanganate (PSM) [2], then the benzaldehydes were condesed with either nitromethane or nitroethene (a large excess of nitroalkane, serving as solvent and reagent, was used) to the corresponding beta-nitroalcohols using either Amberlite IRA-420 or DOWEX-1. Immediately after that the nitroalkenes were formed by esterifying the alcohols with trifluoroacetic acid in DCM, then the nitrostyrenes was formed by reaction with triethylamine.
[2] Polymer supported permanganate (PSM) was prepared by filtering an aqueous solution of potassium permanganate through Amberlyst A-27, subsequent washing of the obtined brick-red material with water and acetone and drying of the beads in vacuo.
Benzyl achohol --> benzaldehyde (95%) --> beta-nitrostyrene (27%)
p-MeO-Benzyl achohol --> p-MeO-benzaldehyde (95%) --> p-MeO-beta-nitrostyrene (23%), p-MeO-beta-methyl-beta-nitrostyrene (25%)
p-NO2-Benzyl achohol --> p-NO2-benzaldehyde (95%) --> p-NO2-beta-nitrostyrene (45%), p-NO2-beta-methyl-beta-nitrostyrene (77%)
p-F-Benzyl achohol --> p-F-benzaldehyde (95%) --> p-F-beta-nitrostyrene (60%)
o-F-Benzyl achohol --> o-F-benzaldehyde (95%) --> o-F-beta-nitrostyrene (65%)
No experimental procedures was given by the assholes
Catalytic hydrogenation freak and the sluttiest slut of all sluts
(Hive Bee)
01-26-03 19:44
No 401639
Thanks for the 1996 Tetrahedron article. When I went up to the chemistry library, the whole year 1996 was *missing*. And as usual, the librarian had no idea what I was talking about...
The J Chem Soc Perkin Trans 1 (1999) article is nice as well, especially for smaller batches (i.e. if you want to organize a drug party with your friends
Ave Hive, synthetisandi te salutant!
(Hive Bee)
12-07-03 04:42
No 475121
(Rated as: excellent)
J. Am. Chem. Soc., 125 (42), 12692 -12693, 2003.
DOI:10.1021/ja0373871
A New Copper Acetate-Bis(oxazoline)-Catalyzed, Enantioselective Henry Reaction
David A. Evans, Daniel Seidel, Magnus Rueping, Hon Wai Lam, Jared T. Shaw, and C. Wade Downey
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138 evans@chemistry.harvard.edu
Received July 18, 2003
Abstract:
A highly enantioselective, nitroaldol reaction catalyzed by a chiral Cu(II) bis(oxazoline) complex has been developed. The reaction scope includes both aromatic and aliphatic aldehydes (15 examples) affording products in good yields and enantioselectivities (87-94% ee). An X-ray structure of the catalyst has been provided along with a rationalization of the sense of asymmetric induction.

The nitroaldol (Henry) reaction is an important carbonyl addition process that affords products that may be transformed into valuable building blocks.1 Therefore, it is not surprising that recent efforts have focused on the development of catalytic enantioselective reaction variants. The current contributions to this area have been highlighted by the recent studies of Shibasaki and Trost.2-5 The purpose of this Communication is to report a new catalyst system for the nitroaldol reaction (eqs 1,2; M = Cu, X = OAc). The basis for the current study was to identify a weakly Lewis acidic metal complex bearing moderately basic charged ligands (X) that would facilitate the deprotonation of nitroalkanes (eq 1) as a prelude to the aldol addition process (eq 2). It was felt that divalent metal acetate-ligand complexes of the general structure A might meet these requirements because acetate has been employed as a Bronsted base in the racemic nitroaldol reaction.1

A series of divalent metal acetates in combination with chiral bidentate ligands were screened as enantioselective catalysts for the nitroaldol process.6,7 From this survey, bis(oxazoline)8 (box) complexes derived from Cu(OAc)2 emerged as promising catalyst candidates. The results from the ligand survey with this metal acetate are summarized in Table 1.
Table 1. Ligand Survey of the Henry Reaction (eq 3)a
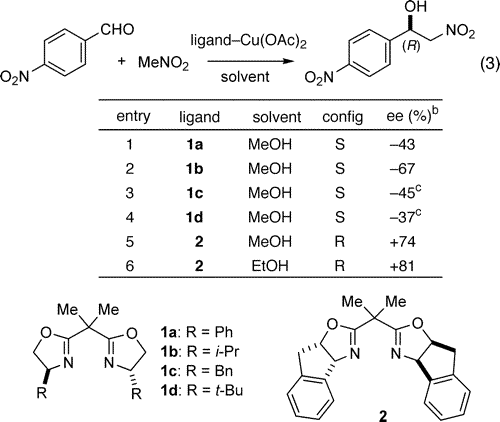
The five box ligands (1a-d, 2) with the illustrated absolute configurations that were evaluated with Cu(OAc)2*H2O afforded promising levels of enantioselection (entries 1-5).8,9 In each instance, the reactions carried out at ambient temperature were complete within 24 h. From this comparison, the indabox ligand 2 proved to be the ligand of choice,9 providing the nitro alcohol product in 74% ee (Table 1, entry 5). With ethanol as the solvent (Table 1, entry 6), the nitro alcohol product was isolated in 81% ee. Further optimization of this process showed that the reaction may be performed with lower catalyst loadings (1-5 mol %), while the use of 10 equiv of nitromethane was found to be sufficient for the reaction to proceed to completion. Reaction concentrations could also be increased to as high as 1.0 M with no change in enantioselectivity. Cu(II) carboxylate structure was also evaluated with ligand 2, and it was concluded that this catalyst variable is subordinate to ligand architecture.10 In all instances, the only side reaction observed in these reactions was the accompanying dehydration product.
With optimized conditions in hand, the scope of the reaction was explored (Table 2). In general, high enantiomeric excesses (87-94% ee) are observed at room temperature for aromatic aldehydes bearing either electron-withdrawing or electron-donating groups (entries 1-9).11 Aliphatic branched and unbranched aldehydes are also acceptable substrates, affording nitro alcohol adducts in good yields and enantioselectivities (entries 10-15, 90-94% ee).
Table 2. Henry Reaction of Nitromethane with Various Aldehydesa

| entry | R | product | time (h) | yield (%)b | ee (%)c |
| 1 | Ph | 3a | 22 | 76 | 94 |
| 2 | 2-MeC6H4 | 3b | 42 | 72 | 93 |
| 3 | 2-MeOC6H4 | 3c | 27 | 91 | 93 |
| 4 | 2-NOC6H4 | 3d | 4 | 86 | 89 |
| 5 | 2-ClC6H4 | 3e | 15 | 88 | 91 |
| 6 | 1-naphthyl | 3f | 15 | 66 | 87 |
| 7 | 4-FC6H4 | 3g | 45 | 70 | 92 |
| 8 | 4-ClC6H4 | 3h | 21 | 73 | 90 |
| 9 | 4-PhC6H4 | 3i | 20 | 70 | 91 |
| 10 | PhCH2CH2 | 3j | 24 | 81 | 90 |
| 11 | i-Bu | 3k | 48 | 86 | 92 |
| 12 | t-Bu | 3l | 96 | 83 | 94 |
| 13 | i-Pr | 3m | 48 | 86 | 94 |
| 14 | n-Bu | 3n | 48 | 87 | 93 |
| 15 | cyclohexyl | 3o | 48 | 95 | 93 |
a All reactions were performed on a 1 mmol scale with 5 mol % of Cu(OAc)2*H2O and 5.5 mol % of ligand 2 at a 0.5 M concentration using 10 equiv of nitromethane in ethanol. Reactions were run at room temperature in a screw-capped vial for the indicated time.
b Values are isolated yields after chromatographic purification.
c Enantiomeric excess was determined by HPLC using Chiracel OD-H, OJ-H, or AD columns.
Reaction enantioselectivity can be further improved by lowering the temperature at the accompanying expense of increasing the reaction time. In one instance, the reaction in entry 1 was performed by mixing the reactants and storing the resulting solution at 0 �C for 10 days. Subsequent isolation afforded the corresponding aldol adduct in 81% yield and 96% ee. When the same reaction was carried out at 40 �C, significant rate acceleration was noted with an accompanying decrease in the enantioselection to 79% and an increase in elimination byproduct.11
Large-scale reactions at minimal catalyst loading (1 mol %) were also evaluated. In one instance, the reaction between 2-methoxybenzaldehyde and nitromethane (as shown in entry 3, Table 2) was performed on a 50 mmol scale with 1 mol % of catalyst loading at a 1.0 M concentration. Following a reaction time of 56 h, the resulting product 3c was isolated in 92% yield (94% ee). Using the same reaction parameters, the nitro alcohol product 3m resulting from a reaction of isobutyraldehyde (as in entry 13) was isolated in 93% yield and 91% ee (108 h reaction time). These results suggest that catalyst loading in the 1% range might well be below the practical limit due to the long reaction times.
The X-ray structure of the chiral copper-ligand complex 4 shown in Figure 1 reveals the expected square planar geometry with the acetate carbonyl moieties oriented toward the vacant apical positions.

Figure 1 Crystal structure of complex 4.
An attempt to rationalize how asymmetric induction is imparted from complex 4 begins with a statement of the impact of the Jahn-Teller (JT) effect on Cu(II) coordination.12 As illustrated in Figure 2, JT distortion of an octahedral Cu(II) complex creates four strongly coordinating and two weakly coordinating sites labeled red and blue, respectively. Addition of a bidentate ligand L2 affords a complex positioning the two cis-oriented strongly coordinating sites in the ligand plane and two trans-oriented weakly coordinating sites perpendicular to the ligand plane (Figure 2, eq 4). For those complexes that simultaneously bind both electrophile and nucleophile, the most reactive transition states should position the nucleophile perpendicular to the ligand plane, while the electrophile, for maximal activation, should be positioned in one of the more Lewis acidic equatorial sites in the ligand plane as illustrated for complex B. By the same argument, complex D should exhibit the lowest reactivity (greatest stability). While transition states B-1 (boat), B-2 (chair), and C-1 (chair) all predict the observed sense of asymmetric induction, our predisposition is to favor B-1 on the basis of both steric and electronic considerations.
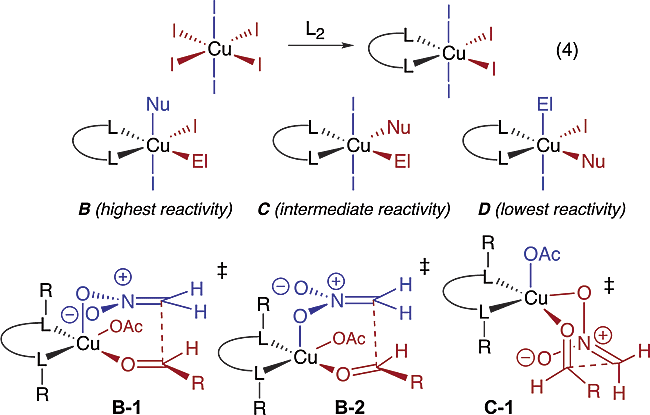
Figure 2 Plausible transition structures for the Henry reaction.
Further investigations into the mechanism and variants of this process are currently underway and will be reported in due course.
Acknowledgment
Support has been provided by the NSF and the NIH (GM 33328-18). D.S., M.R., and H.W.L. gratefully acknowledge Postdoctoral Fellowships from the Ernst Schering Research Foundation, the Deutsche Akademischer Austauschdienst (DAAD), and GlaxoSmithKline, respectively.
Supporting Information Available (http://pubs3.acs.org/acs/journals/suppo
Experimental procedures, spectral data, and stereochemical proofs for all compounds (PDF and CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
* In papers with more than one author, the asterisk indicates the name of the author to whom inquiries about the paper should be addressed.
References
1. For recent reviews, see: (a) Luzio, F. A. Tetrahedron 2001, 57, 915-945. (b) Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: New York, 2001. (c) Seebach, D.; Beck, A. K.; Mukhopadhyay, T.; Thomas, E. Helv. Chim. Acta 1982, 65, 1101-1133.
2. (a) Sasai, H.; Suzuki, T.; Arai, S.; Shibasaki, M. J. Am. Chem. Soc. 1992, 114, 4418-4420. (b) Shibasaki, M.; Yoshikawa, N. Chem. Rev. 2002, 102, 2187-2209 and references therein.
3. (a) Trost, B.; Yeh, V. S. C. Angew. Chem., Int. Ed. 2002, 41, 861-863. (b) Trost, B.; Yeh, V. S. C.; Ito, H.; Bremeyer, N. Org. Lett. 2002, 4, 2621-2623.
4. For related studies, see: (a) Christensen, C.; Juhl, C.; Jorgensen, K. A. Chem. Commun. 2001, 2222-2223.(b) Christensen, C.; Juhl, C.; Hazell, R. G.; Jorgensen, K. A. J. Org. Chem. 2002, 67, 4875-4881.(c) Risgaard, T.; Gothelf, K. V.; Jorgensen, K. A. Org. Biomol. Chem. 2003, 153-156.
5. For enantioselective reactions mediated by chiral quaternary salts, see: (a) Ooi, T.; Doda, K.; Maruoka, K. J. Am. Chem. Soc. 2003, 125, 2054-2055. (b) Corey, E. J.; Zhang, F.-Y. Angew. Chem., Int. Ed. 1999, 38, 1931-1934.
6. Other metal salts were also screened in combination with ligand 2 and methanol as solvent, but were found to be inferior to Cu(OAc)2·H2O: Ni(OAc)2·4H2O, 30% ee; Co(OAc)2·4H2O, 20% ee; Mn(OAc)2·4H2O, 0% ee; Zn(OAc)2·4H2O, 0% ee; Mg(OAc)2·4H2O, 0% ee.
7. Interestingly, CuF2, when used in place of Cu(OAc)2·H2O, gave very similar results (73% ee); however, the reaction rate was significantly lower.
8. Chiral bis(oxazolines) have frequently been used as ligands in a variety of asymmetric processes. For recent reviews, see: (a) Ghosh, A. K.; Mathivanan, P.; Cappiello, J. Tetrahedron: Asymmetry 1998, 9, 1-45. (b) Johnson, J. S.; Evans, D. A. Acc. Chem. Res. 2000, 33, 325-335.
9. Davies, I. W.; Gerena, L.; Lu, N.; Larsen, R. D.; Reider, P. J. J. Org. Chem. 1996, 61, 9629-9630.
10. The influence of carboxylate variable was made on the reaction of PhCHO with nitromethane, using ligand 2 and the indicated Cu(II) carboxylate: Cu(OAc)2·(H2O) (94% ee); Cu(OCHO)2·(H2O) (92% ee); Cu(benzoate)2 (87% ee); Cu(pivalate)2 (92% ee); Cu(2,4-dimethoxybenzoate)2 (96% ee); Cu(4-cyclohexylbutyrate)2 (95% ee).
11. For some substrates (e.g., Table 2, entries 1, 2, and 6), lower yields are observed due to subsequent elimination of the nitro alcohol.
12. (a) Hathaway, B. J. Copper. In Comprehensive Coordination Chemistry; Wilkinson, G., Ed.; Pergamon Press: New York, 1987; Vol. 5, Chapter 53. (b) Hathaway, B. J.; Billing, D. E. Coord. Chem. Rev. 1970, 5, 143-207.
The candle that burns twice as bright burns half as long
(Hive Bee)
03-09-04 07:50
No 493875
again from Kumar, but this time with the 2-nitroalcohol as product instead of nitroalkene. this is better than most of the above rxs on the basis of its speed and low cost of catalyst. no amberlyst or weird complexes here! the yield is only 75%.
the reaction time is 5 minutes, there is no solvent and no side reactions, only unreacted precursors. the catalyst is silica.
they use a fine SiO2 and mention "activation" but give no details. it would be interesting to compare results from ground-up sand, commercial SiO2, dried silica gel, ground-up diatomaceous earth, and SiO2 from home dehydration of homemade (from sand) silicic acid.
the procedure isnt spelled out exactly step-by-step as some bees would want. what was added to what first? what was the exact size of the container? tough.
equimolar amts. of benzaldehyde and nitroethane were adsorbed onto SiO2 in a test tube, placed in the microwave, and cooked for 5 min. (1min. pulses with 30 sec. rests) @ 600W. product extracted with DCM, which was evap'd and product chromatographed for isolation of the 2-nitro-1-phenyl-1-propanol in 75% yield:
SiO2 Catalyzed Henry Reaction: Microwave-Assisted Preparation of 2-nitroalcohols In Dry Media
by Kumar, Reddy, and Yadav
Chemistry Letters vol 27(7), 637-638 1998) (http://www.jstage.jst.go.jp/article/cl/
makes you wonder what else is lurking out there for the experimenter, using things like sand instead of exotics.
(Chief Bee)
03-09-04 08:32
No 493882
I have used that procedure with 4-MeO-PhCHO/MeNO2, and it is OK, but you need to separate the nitroalcohol from byproduct nitroalkene by column chromatography, which might be a hassle for some.
The size of the container or addition order aren't critical, as you adsorb the reactants on silica gel before putting it into the test tube. It is a waste of time trying to use sand/diatomaceous earth, as you really need to use fine silica gel (= activated SiO2) for it to work.
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
03-09-04 10:33
No 493907
thanks for the edit Rhodium, looks much better. my point about procedure was intended as a gentle flame after reading so many posts in lesser forums. about the various forms of 1000 mesh silica, pure, not, crystalline or not, we'll see. you never know for sure unless you try, even if others have. but the main thing- you find the article in error about the lack of side reactions in the real world on a real scale? guess so since thats what you said. someone should kindly volunteer to test this.
in other words, is this article crap that should be flushed?
(Chief Bee)
03-09-04 11:15
No 493916
I merely stated that the nitroalkene invariably formed probably needs column chromatography to be removed. I'd guess that the amount of nitroalkene will not be as large if using less activated benzaldehydes (without methoxy groups).
The Hive - Clandestine Chemists Without Borders
04-05-04 18:59
(Rated as: planned illegal activity)
(Fragile ego)
04-06-04 02:46
No 499257
This will be the first illegal compound I've ever made if I succeed.
Great, But this board is not here to help you make illegal chemicals. Its just for theoretical information exchange.
http://www.democracynow.org/ - The Exception to the Rulers
(Hive Bee)
04-06-04 17:43
No 499355
Apologies, it was just a moment of misplaced bravado. I've never made any illegal compounds and I never intend to. I was just interested in finding out if these chemical reactions worked. I still am.
(Hive Bee)
04-07-04 15:58
No 499517
(Rated as: good read)
I'll assume for the sake of argument that you are going to try this method on an ephedrine analogue which will give a legal analogue.
A literature search gave no hits for the O-acetylation of ephedrine or phenylpropanolamine with acetic acid. Extending the search to other benzylic alcohols gave for 1-phenylethanol to O-acetyl-1-phenylethanol:
Cu(NO3)2*3H2O/AcOH in 90% yield after 2 hours: Synth.Commun.; 28; 11; 1998; 1923-1934.
SiO2/AcOH in DCM giving an 88% yield after 4 hours: Synth.Commun.; 26; 14; 1996; 2715-2722.
Sodium borohydride/AcOH at 85oC - 90oC for 3 hours (unspecified yield): Indian J.Chem.Sect.B; 19; 9; 1980; 822-823.
There were no refs for the sulfuric acid catalysed acetylation until the side chain had been further shortened to phenylmethanol, giving O-acetylphenylmethanol upon treatment with acetic acid/sulfuric acid. For this transformation see:
Justus Liebigs Ann. Chem.; 88; 1853; 130.
Patent DE529135
I don't know whether the absence of references for sulfuric acid catalysed acetylations on longer chains than that of phenylmethanol is an indication of potential dehydration or not, but since the process is an equilibrium when applied to ephedrine derivatives according to Phenyl-2-propanone from Ephedrine Derivatives (../rhodium /phenyl
You don't need to separate the ester from the unreacted alcohol. Once the CTH reduction is over you will be left with any unacetylated ephedrine derivative, whose freebase is soluble in water, and the methamphetamine derivative whose freebase is not. The freebases of the products can also be separated by steam distillation; see Steam volatility of methamphetamine, amphetamine and ephedrine (../rhodium /meth.e
Your assumption that the amino group won't react under the conditions for esterification is not entirely correct, and the side reactions caused by the presence of the amino group is in my opinion the biggest drawback of this otherwise excellent patent. You will notice that with the acetic anhydride/acetic acid O-acetylation used in the original patent that the hydrocholride salt of the phenylpropanolamine derivative is used and, even then, there is some amide formation. You will end up with a majority of O-acetylephedrine analogue, but there will be a small amount each of unreacted ephedrine analogue, N-acetylephedrine analogue, and N,O-diacetylephedrine analogue. Beginning from a vicinal aminoalcohol, as you are, there is probably no way to entirely control the acetylation, even when it is in the non-basic salt form. This is why it Barium got so excited about the possiblilty of acetylating the nitroalcohol, which can't form any acetylated byproduce. But then there is the serious threat of dehydration (for example Post 395584 (Barium: "Oh my!", Novel Discourse)).
If you could instead make your nickel carbonate into nickel bromide, you could try the article posted above by Rhodium: Nickel bromide as a hydrogen transfer catalyst (../rhodium/pdf /cth.nickelbr
The patent in question uses ammonium formate, but sodium formate is a more powerful hydrogen donor. I plan on attempting a modification of this patent (for which I have my own set of questions), and I plan on using potassium formate. A better hydrogen donor should theoretically give better yields, but the problem in this case is the better hydrogen donor is also a stronger base, which may well lead to the acetyl group migrating from the oxygen to the nitrogen - in which case you'll end up with a good yield of pretty useless N-acetylephedrine analogue..
Good luck, and be sure to report back if you have any news.
(Hive Bee)
04-11-04 01:57
No 500139
The idea of O-acetylating the formed nitroalcohol to prevent N-acetylation is a clever one, as is the simultaneous CTH reduction of the O-acetyl and the nitro groups to give the amphetamine. It's unfortunate that under the conditions employed for acetylation, dehydration to the nitrostyrene is a major side-reaction.
If however an azidoalcohol is used, such as 2-azido-1-phenyl-1-propanol, the less electron withdrawing azido group will allow for O-acetylation without the risk of dehydration to the azidostyrene, overcoming this problem.
However, I cannot figure a way around the next potential problem. Assuming that the acetylated nitroalcohol is stable in the same way as the acetylated azidoalcohol is, it would seem both will suffer the same problem upon CTH reduction in any attempt to give the amphetamine.
As the nitro and azido groups reduce readily under the conditions, these will reduce first, leaving the O-acetylated phenylpropanolamine freebase. Even with the amine as its hydrochloride salt, as in the patent, only a very weak base such as the formate salt is sufficient to cause migration of the acetyl group to the nitrogen. This problem will be far worse with the acetylated PPA as freebase, which it will be as it is formed during the course of the reaction. Adding an equivalent of HCl at the beginning of the reaction will of course react with the formate salt instead. From the patent:
Ammonium formate appears to catalyze the rearrangement of O-acetylnorephedrine hydrochloride to N-acetylnorephedrine. Three samples of O-acetylnorephedrine hydrochloride (Experiment Z recrystallized) were stirred with water for six hours respectively at room temperature, at 60 degree C., and at 60 degree C. with ammonium formate added. [...] With the ammonium formate mixture at 60 degree C., the percentage of O-acetylnorephedrine hydrochloride dropped to 91.23% within 30 minutes and to 77.20% by 6 hours while the amount of N-acetylnorephedrine increased from 7.51% to 20.95%.
Is this problem surmountable, or should I instead think of brominating the azidohydrin to the azidobromide and then performing a one-pot CTH to the amine from there? Which will of course present its own potential problems of aziridine formation if nothing else.
The only thing I can think of is to exchange the formic acid salt for formic acid, to which an equivalent of HCl could be added. But I don't know what effect this will have on the yield. Before thinking of this problem I had my heart set on a yield of 90% for a beautiful one-pot reduction...
(Hive Bee)
04-13-04 09:24
No 500517
I see the problem, but I can't imagine how you can deal with it since formic acid works as a CTH hydrogen donor only as a formate anion (I think). That is in buffered conditions that lead to the O- to N-acetyl migration. I don�t know much on CTH but since this is so interesting I will let my imagination go wild so somebee can enjoy using his imagination to correct mine.
Maybe you can use a HCOOK/HCOOH acidic buffer? But I don�t know if it HCOOK would retain its CTH hydrogen donor properties.
Or maybe you can try using an O-formyl-ephedrine hydrochloride (from ephedrine�HCl in refluxing formic acid?) while first adding only a part of potassium formate (maybe 1/4 mol. equivalent). This would form some O-formyl-ephedrine formate (and KCl) which would hopefully bee a hydrogen donor as well as the substrate. The point is that once the reduction starts new formic acid would form neutralizing the basic amino group of the O-formyl-ephedrine or the amphetamine and again forming a hydrogen donor formate (thus replacing the consumed formic acid which lefts the reaction as CO2). I guess this could bee called autocatalysis but who knows if amphetamine can substitute for ammonia or potassium. At the end, while checking the pH, you can slowly add some more potassium formate to keep it going faster and to the end.
I hope I wrote this in an understandable form. The key point is that as soon as a formate anion is consumed a new one forms keeping the pH buffered at the only slightly acidic point due to the constant presence of some O-formyl-ephedrine/amphetamine hydrochloride.
Anyway, maybe it would be better to simply find a hydrogen donor that works in acidic medium. Maybe cyclohexene or tetraline?
Thinking of this I came to a crazy or stupid idea (still haven�t decided on that
�The real drug-problem is that we need more and better drugs.� � J. Ott
(Heavyweight Chempion(eer))
04-13-04 10:30
No 500534
I would try the reduction with 25-50% aq. HOAc as solvent. Voil�, great acidic solvent.
(Hive Bee)
04-13-04 11:37
No 500541
Thanks to both of you for your your replies; I have everything but the 1-(5-indanyl)-2-azidopropane to try this out. I'll give your suggestions a try once I get my hands on some.
While searching for acetylations, I found three references for the acetylation of nitroalcohols with acetic anhydride. The product in all cases is 1-acetoxy-2-nitro-1-phenylpropane:
J. Org Chem; 57; 18; 1992; 4912-4924, giving a 58% yield using acetic anhydride and DMAP in ether;
Bull Chem Soc Jpn; 61; 11; 1988; 4029-4036, using acetic anhydride and pyridine. No yield is given in the abstract;
J. Heterocycl Chem; 31; 4; 1994; 707-710 with acetic anhydride, sulfuric acid in DCM. Again, no yield was given in the abstract.
I should be able to get hold of any of the 3 articles if anyone is interested.
(Heavyweight Chempion(eer))
04-14-04 01:41
No 500683
All three, pretty please.
(Newbee)
04-14-04 02:04
No 500688
Can't get the others...
Inter- and intramolecular [4 + 2] cycloadditions of nitroalkenes with olefins. 2-Nitrostyrenes
Scott E. Denmark, Brenda S. Kesler, Young Choon Moon;
J. Org. Chem.; 1992; 57(18); 4912-4924.
CONSUME * GOVERNMENT IS ON YOUR SIDE * ETHANOL * WATCH TV * DO NOT THINK * BUY THIS PRODUCT
(A Truly Remarkable HyperLab Bee)
04-14-04 19:56
No 500821
Bull Chem Soc Jpn; 61; 11; 1988; 4029-4036, using acetic anhydride and pyridine. No yield is given in the abstract
The article is now placed into an appropriate thread:
Post 500814 (azole: "reduction with Hantzsch ester/SiO2 (full article)", Novel Discourse).
Indeed, acylation of an aliphatic nitro alcohol with Ac2O/Py gave the acetate which was converted into nitroalkene (the target compound) without isolation.
However, acetylation of nitroalcohols derived from aromatic aldehydes is hardly possible due to easy elimination. The product (nitroalkene) will be stabilized by conjugation with the aromatic ring. For examples of such elimination see
Post 369067 (Ritter: "reference", Novel Discourse)
and Post 395584 (Barium: "Oh my!", Novel Discourse).
(Hive Bee)
04-15-04 18:55
No 500975
(Rated as: good read)
The interesting part of it, anyway - interesting being the operative word. The rest of the paper is on the use of the nitroacetates for substituted pyrrole syntheses.
The authors use the method with a number of starting aldehydes. Benzaldehyde and p-chlorobenzaldehyde were the only aromatic aldehydes used. Nitroethane was used in the condensation in the case of both of these aldehydes.
Regioselective Synthesis of 5-Unsubstituted Benzyl Pyrrole-2-carboxylates from Benzyl Isocyanoacetate
Noboru Ono, Hiromi Katayama, Siho Nisyiyama and Takuji Ogawa
Journal of Heterocyclic Chemistry, 31, 707 (1994).
Abstract
A general synthesis of 5-unsubstituted benzyl pyrrole-2-carboxylates was developed based on the reaction of beta-nitroacetates with benzyl isocyanoacetate. The advantage of this route over other pyrrole syntheses was the regiochemical control of the substitution pattern on the pyrrole ring.
General Procedure for the Synthesis of Nitroalcohols and beta-Nitroacetate (2).
To a stirred solution of nitro compounds (10 mmoles) and aldehydes (10 mmoles) in 5 ml of tetrahydrofuran was added DBU (0.1 g) at 10-20�. The resulting mixture was stirred at room temperature for 5-10 hours depending on the substrates. The mixture was then diluted with ether and water. The organic layer was washed with saturated aqueous sodium hydrogencarbonate and brine. The aqueous layers were back-extracted with diethyl ether, dried with magnesium sulfate, filtered, and concentrated in vacuo to afford oils. The crude nitroalcohols were taken up in dichloromethane and treated with acetic anhydride and sulfonic acid (0.1 g). After being stirred for 1-3 hours at room temperature, the reaction mixture was poured into water. The organic layer was washed with aqueous sodium hydrogencarbonate and then concentrated. Purification by short column chromatography (silica gel, hexane-ethyl acetate) gave 2 in 80-90% yield, which was used directly in the next step. Most of the compounds 2 are known except for 2i.
(DBU is 1,8-diazabicyclo[5.4.0]undec-7-ene)
(Hive Bee)
05-03-04 22:07
No 504623
(Rated as: excellent)
Before attempting a simultaneous reduction of both the azido and acetoxy functions of 1-acetoxy-1-(5-indanyl)-2-azidopropane, I decided to test the reduction of 1-acetoxy-2-methylamino-1-phenylbutane to see if the patent worked as described.
This was my first CTH reduction, which may in part explain the yields. But the point is: it works.
It would be great if someone more experienced could give this a try to see how far we can push the yields. The procedure works beautifully and should tolerate far more functional groups (e.g. aryl ethers) than the over-zealous HI/P reduction.
Experimental
1-acetoxy-2-methylamino-1-phenylbutane HCl
9.7g (45mmol) 2-methylamino-1-phenyl-1-butanol HCl1
5.3ml (56mmol) acetic anhydride
Acetic acid
THF
The aminoalcohol HCl was stirred in 20ml acetic acid at 50oC to create a uniform slurry. Acetic anhydride was added in one portion and the temperature was increased to 80oC, by which point the salt had fully dissolved. Stirring was continued at 80-85o for 2 hours then the still hot solution was diluted with 60ml THF,2 causing a heavy precipitate to fall out of solution almost immediately. The flask was allowed to cool to room temperature then left in the freezer overnight. The precipitate was isolated by filtration, rinsed with THF and dried under vacuum.
Yield: 9.2g (35.6mmol, 79%)3
Methaephetamine
9.2g (35.6mmol) 1-acetoxy-2-methylamino-1-phenylbutane HCl
9.0g (107mmol) potassium formate
250mg 10% Pd/C
Water
DCM
Ether
The O-acetoxy compound from the previous step was stirred with 25ml water at 50oC until all had dissolved. The catalyst was added and the temperature was increased to 70oC. A solution of HCOOK in 10ml water was then added over 5 minutes. Gas evolution was noted almost immediately. The reaction was stirred at 70-75oC for 2 hours then allowed to cool. The solution was basic at this point so concentrated HCl was added to bring the pH down to 1. A small amount of celite was added with stirring, then the slurry was filtered and the filtrate washed with 2x50ml DCM. The filtrate was then made strongly basic with NaOH solution and the liberated freebase taken up in 2x50ml ether. The extracts were washed with 2x50ml brine and dried over magnesium sulfate. Removal of the solvent afforded the title product as a clear, colourless oil.4
Yield: 2.9g (18mmol, 51%)5
Comments and references
1 This was the remaining intermediate from Post 475219 (Kinetic: "Benzene -> methaephetamine", Novel Discourse).
2 The precipitate was very fine and difficult to filter. Heptane is recommended in the patent as the precipitate is granular, making this process easier.
3 Some of the product may well still be in solution at this point. It should be possible to remove the acetic acid and excess acetic anhydride under vacuum instead of adding a further solvent to cause precipitation. Unreacted, N-acetylated, or N,O-diacetylated products should not interfere in the CTH reduction and will be removed during the final workup.
4 100mg of which was bioassayed 1 1/2 hours ago. Warm and fuzzy feelings all round.
5 Migration of the acetyl group to the nitrogen is probably the most significant factor in the low yield for the reduction step. According to ../rhodium/pdf /acetylephedri
(Hive Bee)
05-26-04 03:00
No 509615
What can I say, Kinetic, you are a great researcher doing such an interesting work, always making routes so simple and brilliant, what an example...
Can someone be kind to get the papers mentioned in Post 499517 (Kinetic: "Catalytic Transfer Information", Novel Discourse)
Cu(NO3)2*3H2O/AcOH in 90% yield after 2 hours: Synth.Commun.; 28; 11; 1998; 1923-1934.
SiO2/AcOH in DCM giving an 88% yield after 4 hours: Synth.Commun.; 26; 14; 1996; 2715-2722.
Sodium borohydride/AcOH at 85oC - 90oC for 3 hours (unspecified yield): Indian J.Chem.Sect.B; 19; 9; 1980; 822-823.
Don't even have to mention that acid chlorides should work as good as anhydrides in this prep. However, H2SO4 is a nice idea for catalyst, polyphosphoric and tosic should also be given a chance.
I really like this route for getting rid of benzylic -OH, esp for possibilty of use on alkyloxylated ring due to mildness. Good work and well written write-up Kinetic!
'I' am a crowd, obeying as many laws As it has members. Chemically impure Are all 'my' beings.
(A Truly Remarkable HyperLab Bee)
05-26-04 16:55
No 509743
(Rated as: excellent)
For Ganesha:
Sodium Borohydride - Carboxylic Acid Systems - A new Method of Acylation of Alcohols, Phenols & Thiophenols
M. Prashad, V. B. Jigajinni, and P. N. Sharma
Ind. J. Chem., 19B, 822-823 (1980).

A number of alcohols was acylated by heating in a solution of NaBH4 in AcOH or EtCOOH at 85-90 �C. Yields of acylated derivatives were invariably better with primary alcohols in comparison to secondary alcohols. Attempts to acylate tertiary alcohols were unsuccessful. Acylation of phenols and thiophenols was observed at the reflux temperature. A plausible mechanism of this acylation includes the formation of a dehydrating agent (B(OCOR)3) from NaBH4and RCOOH.
(Hive Bee)
06-11-04 21:06
No 512841
(Rated as: excellent)
What can I say, Ganesha; you are a master of the art of flattery. Here are the remaining papers I now feel obliged to provide:
Selective Acetylation of Primary Alcohols: Acetyl and Formyl Transfer Reactions With Copper (II) Salts
N. Iranpoor, H. Firouzabadi and M. A. Zolfigol
Synthetic Communications, 28 (11), 1923-1934 (1998)
Carboxylic Acids Supported on Silica: A Smooth Acylating Agent for Alcohols
Maria da Graca Nascimento, Sandra Patricia Zanotto, Marivania Scremin and Marcos Caroli Rezende
Synthetic Communications, 28 (14), 2715-2721 (1996)
(Hive Bee)
08-01-04 19:48
No 523101
(Rated as: excellent)
The following paper confirms that acetylation of aryl nitroalcohols is indeed possible, and details the acetylation of 1-phenyl-2-nitroethanol to give its O-acetyl ester in 95% yield. The key seems to be the use of acidic catalysis (5-10% MgBr2) which reduces the chance of dehydration:
Magnesium Bromide Catalysed Acylation of Alcohols
Sunil V. Pansare,* Mahesh G. Malusare and Anand N. Rai
Synthetic Communications, 30(14), 2587-2592 (2000)
Abstract
Magnesium bromide is an efficient cataylst for the acetylation and benzoylation of a variety of primary and secondary alcohols with the respective acid anhydrides at ambient temperature. Acetylation of tertiary alcohols requires subambient temperature to suppress competing dehydration. Coordinating solvents retard the acylation process.
(Chief Bee)
08-08-04 20:37
No 524278
(Rated as: good read)
Potassium Fluoride Assisted Selective Acetylation of Alcohols with Acetic Acid
J. W. John Bosco, B. Rama Raju, Anil K. Saikia, Synth. Commun. 34(15), 2849-2855 (2004)
DOI:10.1081/SCC-200026245

Abstract
Potassium fluoride promotes the acetylation of primary and secondary alcohols with acetic acid in excellent yield. Phenols are not affected under this reaction conditions. The groups like double bond, chloro, methoxy, benzyloxy, thiol, and nitro remain unaffected.
It was observed that primary and secondary alcohols can be acetylated readily with a very high yield and neither fluorinated nor eliminated products were identified. This is due to the fact that the hydrogen bonding between fluoride ion and hydroxyl group of acetic acid reduces nucleophilicity and basicity of the fluoride ion. In the case of tertiary alcohol, the reaction becomes sluggish and takes longer time with the formation of elimination products and the reaction ends up with a low yield. However, phenols are not affected and alcoholic group can be selectively acetylated. More importantly, hydroxyl groups residing in chiral center could be acetylated with retention of configuration. The groups like double bond, chloro, nitro, methoxy, benzyloxy, thiol, etc. remain unaffected. Moreover, the reaction does not require any dry glassware and inert atmosphere. The operation is quite simple, because the dehydrating systems like Dean�Stark apparatus or agent like molecular sieve is not necessary. The purification process is very simple, as both potassium fluoride and acetic acid are water-soluble and final product does not require any tedious purification procedures.
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
08-08-04 21:18
No 524285
Kinetic Hoping not to be off topic , my question is on a particular Amino alcohol, Phenylalaninol, once acylated with any of the great methods listed on this thread , many posted by you, the reduction to a hydrocarbon could be ......? I posted some time ago Post 516488 (java: "Removal of Acyl protecting Groups....", Newbee Forum) on de-acylation which may or may not apply here , however I don't know if the reduced product would be the respective hydrocarbon.
Another question would any of this acylation methods acylate the amine group as well as the -OH of the before mentioned alcohol, so that when the reduction occurs the amine would be alkylated and the alcohol reduced to the hydrocarbon?
Maybe the answer is here but just looking too hard...that happens sometimes.......java
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/
(Hive Bee)
08-08-04 22:07
No 524292
Java, before Kinetic answers your question on the reduction, I just want to warn you that the paper you mention in Post 516488 (java: "Removal of Acyl protecting Groups....", Newbee Forum) is about hydrolisys and not about reduction like you seam to imply. That means that you would get your phenylalaninol back from O-acetyl-phenylalaninol and not amphetamine.
BTW, to reduce the OH in phenylalaninol you have to esterify it with an acid that forms a much better leaving group than acetyl (tosyl, halides, mesityl, triflate etc).
�The real drug-problem is that we need more and better drugs.� � J. Ott
(Hive Bee)
08-08-04 23:44
No 524307
I posted some time ago Post 516488 (java: "Removal of Acyl protecting Groups....", Newbee Forum) ....", Newbee Forum) on de-acylation which may or may not apply here , however I don't know if the reduced product would be the respective hydrocarbon.
Nicodem ....you're correct ,sorry if I caused some confusion...
is about hydrolisys and not about reduction like you seam to imply
but pehaps my wishful thinking implied more than just "may or may not apply here".
As to acylating the OH maybe perhaps it would be an easier leaving group that can be hydrogenated with Pd and get the respective hydrocarbon as Kinetic speculated in the application of acylating amino alcohols . I guess I will go back and read the whole thread again to clear this point.
By the way there is a method of removing the acetyl group in Phenyalanine as mentioned in...
Post 523004 (java: "Hydrogenolysis of Phenylalaninol.....", Serious Chemistry)
and methylating the amine group........java
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/