In the course of preparation of a key intermediate for the synthesis of certain anthracycline analogs, we were in need of 6-bromo-2,5-dimethoxybenzaldehyde (3). Rubenstein reported 3 as the exclusive product of the bromination of 2,5-dimethoxybenzaldehyde (1) with bromine in acetic acid1. This assumption was apparently based on the finding* that nitration of 1 gave the 6-nitro and 4-nitro isomers in 80% and 20% yields, respectively.
Later, an isomer of 3, namely 4-bromo-2,5-dimethoxybenzaldehyde (2), was obtained from 1 utilizing anhydrous stannous chloride and bromine in methylene chloride2, since it was believed that 2 was unknown in the literature. Notably, Bortnik et al.3 showed that the structure of the product of the bromination of 2,5-dimethoxybenzaldehyde (1) obtained by Rubenstein was actually 2. These authors further established the identity of this substance by comparing it with the aldehyde 2 obtained in the formylation of the dimethyl ether of bromohydroquinone with hydrogen cyanide and aluminum chloride.

Furthermore, the aldehyde 2 was oxidized to the corresponding known acid and the structures confirmed by 1H-NMR spectral analysis. Although the original incorrect assignment of the structure reported by Rubenstein1 has since been corrected by several investigators2-5, the regioisomer 3 was never identified from the reaction of 1 with bromine in acetic acid. We have repeated the bromination of 1 as described by Rubenstein and now report our findings.
The action of bromine on 2,5-dimethoxybenzaldehyde (1) in glacial acetic acid at room temperature afforded a mixture of two crystalline products, differing slightly in polarity, as indicated by thin layer chromatographic analysis. The separation of this mixture was achieved by fractional recrystallization (95% ethanol) and column chromatography to obtain 2 and 3. The 1H NMR spectrum of 3 displayed an AB pattern for two protons in the aromatic region (δ = 7.00 ppm, dd, J = 9 Hz). The desired product 3, was obtained in only 5% yield in contrast to 2 which comprised the major portion (87%) of the reaction product. The structure 2 was assigned based on 1H NMR spectral analysis and comparison with literature data2,3. Final proof of the position of the bromine atom in the regioisomers 2 and 3 came from the subsequent synthesis of 3,6-dimethoxybenzocyclobutene (9) via Parham cyclialkylation6 of 8 (Scheme 1). Thus, the treatment of 3 with triphenyl methoxymethylphosphonium chloride in the presence of sodium ethoxide in ethanol7 at 65°C resulted in a mixture of products. Among the products isolated were trans-6-bromo-2,5-dimethoxy-βmethoxystyrene (4) (64%)8, mp 66-67°C, and its cis isomer 5 (4%), 8 mp 92-95°C, and 2-bromo-3,6dimethoxyphenylacetaldehyde (6) along with a trace of unreacted 3. The structural assignment for trans and cis compounds was made on the basis of coupling constant values exhibited by the olefinic protons (Jtrans = 13 Hz vs. Jcis = 6 Hz).
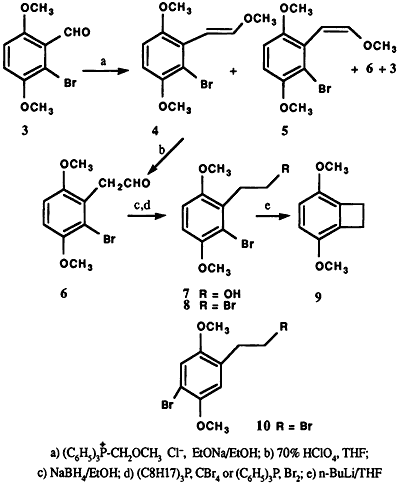
Hydrolysis of the E-Z mixture (4,5) with 70% perchloric acid in tetrahydrofuran9,10 at room temperature furnished 6 in 98% yield. Reduction of the aldehyde 6 with sodium borohydride in 95% ethanol at 50°C afforded the alcohol 7 (86%). Conversion of the bromo alcohol 7 to the corresponding bromide 8 (62%) was effected utilizing tri-n-octylphosphine and carbon tetrabromide11 or triphenylphosphine dibromide12. Finally, lithium halogen exchange of 8 with n-butyllithium in dry tetrahydrofuran (-105°C to -95°C), followed by intramolecular cyclization upon warming, provided the benzocyclobutene 9 (Scheme 1)13. A similar synthetic sequence starting from the bromo isomer 2 resulted only in the isolation of the dehalogenated end products, as would be expected, upon attempted cyclization of 10, mp 75-76°C (EtOH)14. Only the regioisomer 8 with bromine atom ortho to the bromoalkyl side chain would readily undergo intramolecular cyclization, following the reaction conditions used, to form the cyclic product 9.
Thus, it is further confirmed that the structure of the minor product of the bromination of 1, following the method reported by Rubenstein1, is 3, while the major product2-5 is indeed 2. To our knowledge, the minor isomer 3, isolated from the reaction mixture, is reported here for the first time.
Experimental
Bromination of 2,5-Dimethoxybenzaldehyde
A cold solution of 20.0 g (0.12 mol) of 2,5dimethoxybenzaldehyde in glacial acetic acid (115 mL) was treated with 20.0 g of bromine in glacial acetic acid (60 mL). The solution was stirred at room temperature for 2-3 days and diluted with ice water. The yellow precipitate was collected by filtration and dried (28 g, 95%), mp 118-126°C (lit.1 mp 125-126°C). Recrystallization from ethanol gave 15.2g of 2, mp 132-133°C (lit.2,3 mp 132-133°C). Ethanol was removed from the mother liquor in vacuo and the residue was subjected to column chromatography (silica gel, benzene). The solid (10.4 g) collected in initial fractions was 4-bromo-2,5-dimethoxybenzaldehyde (2), total yield 25.6g (87%), mp 132-133°C. Subsequent fractions gave 1.5g (5%) of 6-bromo-2,5-dimethoxybenzaldehyde (3), mp 102-103°C (EtOH).
6-Bromo-2,5-dimethoxy-beta-methoxystyrenes (4,5)
To a solution of sodium ethoxide (prepared from 0.15 g, 6.61 mmol of sodium in 10 mL of absolute ethanol) was added 1.78 g (5.19 mmol) of triphenylmethoxymethylphosphonium chloride7. The mixture was stirred at room temperature for fifteen minutes. To this mixture was added 1.27 g (5.18 mmol) of 6-bromo-2,5-dimethoxybenzaldehyde (3) in portions. The reaction mixture was stirred at room temperature for ten minutes followed by 24 hrs at 65°C in an argon atmosphere. After cooling, the reaction mixture was poured over ice water and repeatedly extracted with ether. The ether extract was washed with water and dried (Na2SO4). Removal of the solvent gave a yellow viscous oil which was chromatographed (silica, benzene). Initial fractions contained some triphenylphosphine. Subsequent fractions gave trans-6bromo-2,5-dimethoxy-β-methoxystyrene (4), (0.92g; 64%), mp 66-67°C. cis-6-bromo-2,5-dimethoxy-β-methoxystyrene (5), (0.060 g, 4%), mp 92-95°C. β-(6-bromo-2,5-dimethoxyphenyl)acetaldehyde (6), (0.12 g, 9%) (analytical data similar to the structure described below). In addition, a trace amount of unreacted aldehyde 3 (10 mg), triphenylphosphine and triphenylphosphineoxide were isolated. Due to apparent instability during recrystallization, the stereoisomers 4 and 5 were hydrolyzed to 6 which was characterized as its 2,4dinitrophenylhydrazone.
beta-(4-Bromo-2,5-dimethoxyphenyl)acetaldehyde (6).
The E-Z mixture (4,5) (0.7 g, 2.56 mmol) in tetrahydrofuran (40 mL) was treated with 6 mL of 70% perchloric acid. After stirring at room temperature for twenty minutes, the solution was poured over crushed ice and extracted with ether. The ether layer was washed with water and dried (MgSO4). Removal of the solvent gave a pale yellow solid (0.65 g, 98%), mp 67-69°C. It was analyzed as its 2,4-dinitrophenylhydrazone, mp 195-196°C (EtOH-EtOAc).
beta-(6-Bromo-2,5-dimethoxyphenyl)ethanol (7)
A solution of 6 (600 mg, 2.32 mmol) in 40 mL of 95% ethanol at 50°C was treated with sodium borohydride (40 mg). The reaction mixture was stirred at room temperature for forty-five minutes. The solvent was removed in vacuo and the residue was treated with hydrochloric acid (10%) and extracted with ether. The ether layer was washed successively with water, sodium bisulfite (10%), sodium carbonate (10%), and dried (MgSO4). Removal of the solvent gave a white solid (0.52 g, 86%), mp 112-113°C (benzene-hexane).
beta-Bromo-2,5-dimethoxyphenydiethyl bromide (8)
A solution of bromoalcohol 7 (86 mg, 0.33 mmol) in dry carbon tetrachloride (3 mL) was added dropwise to a suspension of triphenylphosphine dibromide (prepared from 250 mg of triphenylphosphine and 160 mg of bromine in 10 mL of carbon tetrachloride). The mixture was stirred for one hour at room temperature and then refluxed for seven hours in an inert atmosphere. After cooling, the reaction mixture was filtered and the filtrate was evaporated in vacuo. The residue was chromatographed (silica, 50% heptane in chloroform) to obtain the dibromide 8 (66 mg, 62%), mp 68-69°C (hexane).
3,6-Dimethoxybenzocyclobutene (9)
In a 10 mL three-neck flask compound 8 (0.194 g , 0.60 mmol) was dissolved in tezrahydrofuran (5 mL, dry, freshly distilled) and hexane (1 mL, dry) in an argon atmosphere. The solution was cooled (-95°C to -105°C). To this solution was added n-butyllithium (0.45 mL, 1.55M in hexane) at such a rate that the internal temperature did not exceed -95°C After stirring for one-half hour (-100°C to -105°C), the reaction mixture was warmed to room temperature and poured into ice water and extracted with ether. The ether layer was washed with water and dried (MgSO4). The residue, obtained after evaporation of the solvent, was subjected to preparative layer chromatography (silica gel, 50% heptane in chloroform). The band at Rf 0.4 gave 9 (0.064 g, 65%); mp 59-60°C (EtOH-H2O).