Reduction of Nitriles to Primary Amines with Nickel Boride at Ambient TemperatureSynth. Comm. 32(8), 1265-69 (2002)[ Back to the Chemistry Archive ]Reduction of a variety of nitriles to their corresponding primary amines can be achieved with nickel boride generated in situ in dry ethanol at ambient temperature. The reductions are very rapid and chemoselective. Sodium borohydride is a milder reducing agent than lithium aluminium hydride (LAH), is comparatively inexpensive, easier to handle and can also be employed in protic solvents [2a-b]. Whereas LAH offers little selectivity in reductions, sodium borohydride does not normally reduce groups like ester, nitro, amides and nitriles. The reducing behaviour of sodium borohydride undergoes a drastic change with the addition of transition metal salts [3a-e]. Among these, Ni(II) salts have drawn wide attention in the modification of sodium borohydride reactivity [4]. Nickel boride, prepared in situ, was initially used as a catalyst for various reductions under hydrogen atmosphere. Nickel boride has been reported as a catalyst in the reduction of nitriles under hydrogen atmosphere [5]. The yields are very poor even after prolonged reaction times e.g., only 56% of benzonitrile was reduced under hydrogen atmosphere (30 psi) after 48 h using nickel boride as catalyst. The focus has shifted more recently to its applications as a reducing agent rather than as a catalyst [6a-d]. Nickel boride, prepared under different conditions, is reported to behave differently in its reactions. Recently Caddick et al. [7] have reported that nitriles are reduced to secondary amines with nickel boride generated in situ from nickel (II) chloride and sodium borohydride. Primary amines can be obtained only as protected primary amines by adding trapping agents like acetic anhydride and di-tert-butyl bicarbonate. We report herein a simple and convenient procedure for the reduction of the robust cyano group in aromatic nitriles 1 to primary amines 2 directly in high yields with nickel boride at ambient temperature. The nickel boride was generated in situ from dry nickel(II) chloride and sodium borohydride and the reductions were carried out in dry ethanol. The reductions are very rapid and are complete in ~5 min, as monitored by thin layer chromatography, using 1:1:3 molar ratio of substrate to nickel(II) chloride to sodium borohydride, Eq. (1). 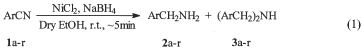
Our results are summarised in Table 1. Reductions carried out with hydrated NiCl2 and/or 95% ethanol gave lower yields of the corresponding primary amines. There was very little reduction of benzonitrile with sodium borohydride alone even after 24 h and also benzonitrile was recovered unchanged when treated with nickel(II) chloride only. The reductions are thus undoubtedly proceeding due to the involvement of both reagents i.e., the nickel boride formed in situ. Ethanol is the solvent of choice and primary amines are obtained by a simple work-up procedure. Reductions were slower in tetrahydrofuran and dimethylformamide, and also yielded a complex mixture with lower yields of the corresponding primary amines. Small amounts of secondary amines 3 were also formed in the reactions. The formation of secondary amines could not be eliminated completely despite attempting reactions under different conditions.8 The secondary amines were formed from the primary amines obtained initially as confirmed by an independent reaction of benzyl amine with nickel boride. However, formation of small amounts of secondary amines by other pathways also cannot be ruled out. Nickel boride loses its activity with time since benzonitrile (1a) and p-chlorobenzonitrile (1d) were recovered unchanged (2h) when treated with a preformed solution of nickel boride after 72 h. The reductions are chemoselective as halo (chloro and bromo), methoxy, dimethylamino, olefinic and naphthyl groups were unaffected under our reaction conditions. It is thus obvious again that reaction products of nickel boride reductions can be varied by changing stoichiometries and/or reaction solvent and temperature.
ExperimentalDry EtOH was prepared by the known procedure. NaBH4 (E. Merck) was used in all the reactions. NiCl26H2O (Qualigens) was used after drying in a nickel crucible in an oven at ~250°C until it turned golden yellow. It was powdered and stored in a vacuum desiccator. Nitriles 1a, 1b and 1r were obtained from commercial sources and were distilled before use. Nitriles 1c–q were prepared from their corresponding aldehydes by treatment with hydroxylamine hydrochloride according to the reported procedure.9 The IR spectra were recorded on a Perkin Elmer FT-IR spectrum 2000 and NMR spectra were recorded on Hitachi FT-NMR model R-600 (60 MHz) with TMS as the internal standard. Mass spectra were recorded on a Jeol JMS D 300 spectrometer using the electron impact method (70 eV). In a typical procedure, in a 100 ml conical flask mounted over a magnetic stirrer, fitted with reflux condenser and a mercury trap, was placed a mixture of nitrile (9.8 mmol), anhyd. nickel (II) chloride (9.8 mmol) and dry ethanol (20 ml). Sodium borohydride (29.4 mmol) was added very cautiously while stirring the solution vigorously. The progress of the reaction was monitored by TLC using benzene : ethyl acetate (90 : 10, v/v) as eluent. After the complete disappearance of the nitrile (~5 min), the reaction mixture was filtered through a celite pad after ~15 min. The filtered nickel boride precipitate was washed with ethanol (2×10 ml). The combined filtrate was diluted with water (150 ml) and extracted with ethyl acetate (3×10 ml). The combined extract was dried over anhyd. MgSO4, filtered and concentrated on a rotary vacuum evaporator. After drying under vacuum, the product was purified by column chromatogaphy over silica gel (20×2 cm; 100–200 mesh) using benzene : ethyl acetate as the eluent. Eluate was concentrated under reduced pressure to afford the products which were analysed by IR, NMR and mass spectra as most of these are known products [10a-b]. References [1] The work was presented at Annual Convention of Chemists. Haridwar, India, Nov. 2000, Handbook, pp. C4.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||