02-03-03 05:12
No 403852
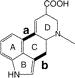
Synthesis of Lysergic Acid Derivatives by Tandem Radical Cyclisation Reactions
Full-text: Synlett 357-358 (1993) (http://pharmacist8.tripod.com/erg-rad-c
Abstract:
A double radical cyclisation of a 2-bromoaniline derivative, initiated with tri-n-butyltin hydride, to construct the lysergic acid ring system is described; formation of a 6- membered D ring is controlled by an intramolecular thermal cyclisation prior to radical addition.
_______ ____ ___ __
See also:
Post 402816 (pHarmacist: "Cobalt-Catalyzed Total Synthesis of (±)-LSD", Tryptamine Chemistry)
Synthetic Studies on (+)-Lysergic Acid (http://www.f.u-tokyo.ac.jp/~fukuyama/sl
Lot of refs...
Note the paper published by R. B. Woodward (1954), according to many the most complete org. chemist of all times and his total synth of L. Acid. He was the first to announce the tot synth of it.
Can someone fetch that paper, Rhodium, lugh? R. B. Woodward et al., J. Am. Chem. Soc., 76, 5256 (1954)
I would love to read it, that man is my absolute and only idol!
Accept No Imitations, There Can Only Bee One; www.the-hive.ws
(Chief Bee)
02-05-03 20:48
No 404782
(Rated as: excellent)
This seems to be a preliminary communication, there must be another more in-depth paper:
R. B. Woodward et al., J. Am. Chem. Soc., 76, 5256 (1954) (../rhodium/pdf /lysergic.woo
This one seems to be somewhat more comprehensive, being 28 pages:
R B Woodward - Total Synthesis of Lysergic Acid - JACS 78, 3087 (1956) (../rhodium/pdf /lysergic.aci
Now also available in HTML at Rhodium.ws (../rhodium /lyserg
I haven't posted this already, right? It's a french article on the total synthesis of L'Acide Lysergique...
Une Nouvelle Synthese De L'Acide Lysergique
M. Julia, F. Le Goffic, J. Igolen & M. Baillarge
Tetrahedron Letters 20, 1569-71 (1969) (../rhodium/pdf /lysergique.p
(Hive Bee)
03-02-03 02:32
No 412911
(Rated as: excellent)
Total Synthesis of (±)-Lysergic acid - A new Diels Alder Pyridine synthesis
Jian Wang
Diss. Abstr. Int. B 62(12), 5740 (2002) (../rhodium/pdf /lysergic.die
Got democracy?http://www.dhushara.com/book/multinet/de
(Hive Bee)
12-13-03 02:09
No 476514
(Rated as: excellent)
Org. Lett., ASAP Article
DOI:10.1021/ol0354369
Web Release Date: December 10, 2003
A New Synthesis of Lysergic Acid
James B. Hendrickson and Jian Wang
Department of Chemistry, Brandeis University, Waltham, Massachusetts 02454-9110
Received July 30, 2003
Abstract:

(±)-Lysergic acid (1) has been synthesized via an economical 8-step route from 4-bromoindole and isocinchomeronic acid without the need to protect the indole during the synthesis. Initial efforts to form the simpler 3-acylindole derivatives first and then cyclize these were unsuccessful in the cyclization step.
The ergot alkaloids pose an unusual opportunity for synthesis. The central alkaloids are all amides of lysergic acid (1) and all possess a broad range of pharmacological activity.1-3 Only one of these, the diethylamide (LSD), however, is strongly and notoriously psychoactive. As such it is listed as a class I controlled substance. Since both the natural and synthetic derivatives are easily convertible to lysergic acid and so to its diethylamide, all of these are also controlled substances. As a result this potential pharmacological treasure is essentially unavailable for practical clinical testing.
We considered that a derivative of lysergic acid bearing an unremoveable substituent, like an added C-alkyl group, could not be converted to lysergic acid itself or its amide. Such derivatives would probably retain the broad pharmacological activity of the ergot family but might easily avoid the unique hallucinogenic property of LSD. This idea encouraged us to seek a short, practical synthesis route to lysergic acid suitable for incorporation of C-alkyl starting materials to create these derivatives.
Lysergic acid has already been synthesized about eight times.4 The shortest path has 11 steps and none are serious candidates for practical manufacture. Every synthesis to date contains redundant protection/deprotection sequences, often as indole starting materials reduced and acylated, only at the end reconstituted to the indole form. To eliminate this redundancy we decided that the indole should be carried through intact.
The simplest convergent bondset for assembling the lysergic skeleton should be the boldface bonds in ring C, which just arise from indole and nicotinic acid starting materials. No previous syntheses had utilized this approach except the Julia route, which did not carry the indole moiety through unchanged.
Of the two initial constructions necessary for the bondset in Figure 1 we began with the simplest (bond b) via an acylation of indole or 4-haloindoles5 2 with the acid chloride 3 from the commercially available 6-carboxynicotinic acid (isocinchomeronic acid), as outlined in Scheme 1. The acid was esterified, selectively hydrolyzed only at the 6-position6 with aqueous Cu(NO3)2, and converted (SOCl2) to 3. The Grignard reagents from 2 were acylated with 3 to form 4. However, a number of attempts at palladium-catalyzed cyclizations of 4 (X = Br or I) or reduced pyridine derivatives of 4 and their N-methyl salts were all unsuccessful.
Figure 1 Bondset of lysergic acid synthesis.
 (http://pubs.acs.org/isubscribe/journals
(http://pubs.acs.org/isubscribe/journalsScheme 1. Attempt via Bondset ba Approacha a
 (http://pubs.acs.org/isubscribe/journals
(http://pubs.acs.org/isubscribe/journalsReagents: (i) EMgBr/ZnCl2/Et2O.
We also considered that a thermal pericyclic cyclization of the anion 5 of 4 might be accessible with subsequent loss of HX to form 6, but several trials of this at elevated temperatures, with or without added base, led only to intractable tars.
The alternative path to close ring C, making bond a first, was ultimately successful, as summarized in Scheme 2. For this approach we needed a nicotinic acid derivative with a halogen marking the 5-position.
Scheme 2. Synthesis of Lysergic Acid via Bondset ab Approacha a
 (http://pubs.acs.org/isubscribe/journals
(http://pubs.acs.org/isubscribe/journalsReagents: (i) Pd(PPh3)4/Na2CO3(aq)/EtOH; (ii) NaBH4/CaCl2/EtOH; (iii) MnO2/CHCl3; (iv) NaOH/MeOH; (v) NaBH4/TFA/CH2Cl2; (vi) MeI/CH2Cl2; (vii) NaBH4/MeOH; (viii) NaOH /EtOH.
The common introduction of halogen on pyridines, via SOCl2 on the N-oxide,7 provides only the ortho/para halides. However, sulfonyl halides can give rise to meta substitution8 and the reaction of the N-oxide of 6-carboxynicotinic acid with thionyl chloride affords9 the m-chloro derivative 7 on workup with methanol. We believe this results from first forming the normal, nonplanar p-chloro intermediate in Figure 2, which can then collapse via the pericyclic rearrangement shown and subsequent loss of the p-chloride to afford 7.
Figure 2 Proposed mechanism for formation of 7.
 (http://pubs.acs.org/isubscribe/journals
(http://pubs.acs.org/isubscribe/journalsWhen the 4-haloindoles 2 were converted to the boronic acid 8 (via KH + BuLi and B(OBu)3), the Suzuki coupling was successful in forming 9a in 91% yield. While this work was in progress, a closely related reaction appeared in a note by Doll10 coupling 8 with 5-bromonicotinic ester, but the subsequent addition of the missing carbon 4 for lysergic acid failed.
We presumed that an appropriate base would easily initiate the cyclization of the diester 9a to the tetracyclic ketone corresponding to 10. However, treatment of the diester with NaH, even in glycol at 197 °C, yielded only starting material. A number of attempts to cyclize the corresponding, very insoluble diacid with thionyl chloride or variants of PPA led only to recovery of diacid or intractable mixtures with no evidence of cyclization.
Following a similar but intermolecular version from Potier11 we reduced the ester selectively12 with NaBH4 and CaCl2 to 9b and then oxidized it to the aldehyde 9c with MnO2. The aldehyde cyclized at room temperature easily and quantitatively with only 2 mol % of NaOH to yield 10. Various efforts to cyclize the alcohol 9b or its tosylate to 11 all recovered only starting materials. The difference in the ease of cyclization of 9c over its precursors surprised us but we became convinced from models that there was severe steric resistance to the stereoelectronic demands for cyclization except for the aldehyde case.
Typical reduction13 of the indole-alcohol 10 with NaBH4/TFA afforded 11, which proved to be unstable, decomposing in a matter of hours. Accordingly, the remaining steps were carried through without isolation. Freshly prepared 11 was methylated directly with methyl iodide and the crude salt 12 reduced with excess NaBH4 in methanol to a mixture of methyl lysergate and its cis-isomer isolysergate in a 6:1 ratio, as a pale yellow solid.
These diastereomers are reported to be somewhat unstable14 and so were immediately hydrolyzed to lysergic acid with NaOH, which also equilibrated them to the more stable lysergic acid, which was then finally recrystallized to mp 241-242 °C (lit. 242-243 °C).
The 1H NMR spectra of the mixed esters were identical with spectra kindly provided by Prof. Ichiya Ninomiya, and the NMR spectra (1H and 13C) of the lysergic acid agreed with that of a natural sample kindly provided by Dr. David Nichols.
The last three operations (10 to 1) are carried out easily in good yield without isolation and purification; this result lends value to the initial conception in Figure 1 that the most economical synthesis of lysergic acid is one that originates in the two main starting materials, a simple indole and a nicotinic acid derivative, both retaining their aromaticity to the very end. This synthesis comprises eight steps from isocinchomeronic acid and 4-bromoindole and proceeds in an unoptimized overall yield of 10.6%. Chirality is only introduced in the final reduction step, and enantioselective measures for this reduction have not yet been examined, nor has the parallel synthesis of C-alkyl derivatives.
Acknowledgment
We thank Dr. Toby Sommer at Brandeis University for his constant help and inspiring discussion, Brandeis University for financial support of this study, Prof. Ninomiya at Kobe Pharmaceutical University, Japan for the comparison of NMR spectra and Dr. David Nichols at Purdue University for a natural sample.
Experimental details and spectral data for new compounds. This material is available free of charge via the Internet at http://pubs3.acs.org/acs/journals/suppor
1. Hofmann, A. Ergot Alkaloids, Pharmacology; Spano, P. F., Trabucchi, M., Eds.; Karger: Basel, Switzerland, 1978; Vol. 16, Supp. 1, p 1.
2. Lemberger, L. Fed. Proc. 1978, 37, 2176.
3. Berde, D.; Schild, O. Handbook of Experimental Pharmacology; Springer Verlag: Berlin, Germany, 1978; Vol. 49, p 1.
4. (a) Kornfeld, E. A.; Fornefeld, E. J.; Kline, G. B.; Mann, M. J.; Morrison, D. E.; Jones, R. G.; Woodward, R. B. J. Am. Chem. Soc. 1956, 78, 3087. (b) Julia, M.; LeGoffic, F.; Igolen, J.; Baillarge, M. Tetrahedron Lett. 1969, 20, 1569. (c) Armstrong, V. W.; Coulton, S.; Ramage, R. Tetrahedron Lett. 1976, 47, 4311. Ramage, R.; Armstrong, V. W.; Coulton, S. Tetrahedron 1981, 9 (Suppl.), 157. (d) Oppolzer, W.; Francotte, E.; Baettig, K. Helv. Chim. Acta 1981, 64 (2), 478. (e) Rebek, J., Jr.; Tai, D. F. Tetrahedron Lett. 1983, 24 (9), 859. Rebek, J., Jr.; Tai, D. F.; Shue,Y. K. J. Am. Chem. Soc. 1984, 106 (6), 1813. (f) Kurihara, T; Terada, T.; Yoneda, R. Chem. Pharm. Bull. 1986, 34 (1), 442. Kurihara, T.; Terada, T.; Harusawa, S.; Yoneda, R. Chem. Pharm. Bull. 1987, 35 (12), 4793. (g) Kiguchi, T.; Hashimoto, C.; Naito, T.; Ninomyia, I. Heterocycles 1982, 19 (12), 2279. Ninomyia, I.; Hashimoto, C.; Kiguchi, T; Naito, T. J. Chem. Soc., Perkin Trans. 1 1985, 941. (h) Cacchi, S.; Ciattini, P. G.; Morera, E.; Ortar, G. Tetrahedron Lett. 1988, 29 (25), 3117.
5. Somei, M.; Kizu, K. Chem. Pharm. Bull. 1985, 33, 3696.
6. Ooi, G. K. S.; Magee, R. J. J. Inorg. Nucl. Chem. 1970, 32, 3315.
7. Joule, J. A.; Miller, K. Heterocyclic Chemistry, 4th ed.; Blackwell Science: Oxford, UK, 2000; p 100
8. Katritzky, A. R.; Lagowski, J. M. The Chemistry of the Pyridine N-Oxides; Academic Press: New York, 1971; p 288.
9. Quarroz, D. Swiss Patent CH 657,124, 1986; Chem. Abstr. 1987, 106, 32852.
10. Doll, M. K.-H. J. Org. Chem. 1999, 64, 1372.[Full text - ACS] (http://pubs.acs.org/cgi-bin/citation?jo
11. Potier, P.; Langlois, Y. Tetrahedron 1975, 31, 419.
12. Matsumoto, I.; Yoshizawa, J. Japanese Patent JP 48029783, 1973.
13. Gribble, G. W.; Nutaitis, C. F. Org. Prep. Proced. Int. 1985, 17, 317.
14. (a) Glenn, A. L. Quart. Rev. 1954, 8, 192. (b) Stoll, A.; Petrzilka, T. Helv. Chim. Acta 1953, 36, 1125.
The candle that burns twice as bright burns half as long
(Chief Bee)
01-03-04 21:36
No 480281
(Rated as: excellent)
The article posted by Lego above has now been officially published in Org. Lett. and can now be found in HTML on my page:
A New Synthesis of Lysergic Acid
James B. Hendrickson and Jian Wang
Organic Letters 6, 3-5 (2004) (../rhodium /lyserg
Does the name James B. Hendrickson ring a bell? This is the professor who used the synthesis planning software SYNGEN to come up with this novel LSD route (it just took quite a while for him to go from the synthons suggested by the program to a workable Lysergic Acid synthesis. Related: Post 14858 (Lilienthal: "Lysergic acid total syntheses / SYNGEN", Tryptamine Chemistry) and Post 211915 (Rhodium: "Re: Somebody Get a Password!!!!", General Discourse)
Also, the co-author Jian Wang, he seems to be the one who wrote the PhD dissertation linked in Post 412911 (ChemisTris: "Total synthesis of (±)-Lysergic acid", Tryptamine Chemistry).
Here below follows some syntheses of LSD and analogs from Lysergic Acid (of which three are written by D.E. Nichols):
____ ___ __ _
Emetic activity of reduced lysergamides
Fatima N. Johnson, Istvan E. Ary, David G. Teiger, Ronald J. Kassel
J. Med. Chem. 16(5), 532-537 (1973) (http://)
Abstract
A new efficient method for the direct amidation of d-lysergic acid was used to prepare a variety of lysergamides. A pharmacological evaluation of these compounds, their di- and tetrahydro derivatives, and derivatives bearing substituents in the indole portion of the molecule showed that, in general, only 9,10-dihydrolysergamides of primary amines possess activity comparable to the potent emetic activity of the components of dihydroergotoxine.
____ ___ __ _
Stereoselective LSD-like activity in d-lysergic acid amides of R- and S-2-aminobutane
Robert Oberlender, Robert C. Pfaff, Michael P. Johnson, Xuemei Huang, David E. Nichols
J. Med. Chem. 35(2), 203-211 (1992) (../rhodium/pdf /nichols/nich
Abstract
The (R)- and (S)-2-butylamides of d-lysergic acid were prepared and evaluated in behavioral and biochemical assays of 5-HT2 agonist activity. In rata trained to discriminate 0.08 mg/kg LSD tarbate from saline, both isomers completely substituted for the training stimulus. Similarly, both isomers were found to possess very high affinity in displacing [125I]-(R)-DOI ([125I]-(R)-1-(2,5-dimethoxy-4-iodophenyl)-2-amino
____ ___ __ _
Synthesis and LSD-like discriminative stimulus properties in a series of N(6)-alkyl norlysergic acid N,N-diethylamide derivatives
Andrew J. Hoffman, David E. Nichols
J. Med. Chem. 28(9), 1252-1255 (1985) (../rhodium/pdf /nichols/nich
Abstract
A convenient method for the synthesis of N(6)-alkyl norlysergic acid N,N-diethylamide derivatives was developed. A series of these compounds was synthesized and tested for substitution in the two-lever drug discrimination assay, in rats trained to discriminate injections of d-LSD tartrate (185.5 nmol/kg, ip) from saline. A dose-response curve for each of the compounds in the series was generated. Structure-activity relationships were developed, based on comparison of the estimated ED50 values from these curves. Of the compounds that substituted for LSD, the N(6)-ethyl and -allyl were approximately 2-3 times more potent than LSD itself. The N(6)-propyl was equipotent to LSD, while the isopropyl derivative was half as active. The n-butyl compound was 1 order of magnitude less potent than LSD, suggesting a similarity to the SAR of certain serotonin and dopamine agonists. By contrast, no generalization occurred to norlysergic acid N,N-diethylamide and the N(6)-2-phenethyl derivative.
____ ___ __ _
Drug Discrimination and Receptor Binding Studies of N-Isopropyl Lysergamide Derivates
Huang, Marona-Lewicka, Pfaff, Nichols
Pharmacology, Biochmistry and Behavior 47(3), 667-673 (1994) (../rhodium/pdf /nichols/nich
Abstract
Isopropyl (IPLA), N-methyl-N-isopropyl (MIPLA), N-ethyl-N-isopropyl (EIPLA), and N,N-diisopropyl (DIPLA) lysergamides were evaluated for lysergic acid diethylamide (LSD)-like activity. In rats trained to discriminate 0.08 mg/kg LSD tartrate from saline, each of the subject compounds completely substituted, with an ED50 two to three times larger than that of LSD except for DIPLA, which had an ED50 about eightfold greater. Similarly, all the compounds displaced [125I](R)-1-(2,5-dimethoxy-4-iodophenyl)-2-a
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
08-06-04 22:36
No 524036
(Rated as: excellent)
Enantioefficient Synthesis of -Ergocryptine: First Direct Synthesis of (+)-Lysergic Acid
Istvan Moldvai, Eszter Temesvari-Major, Maria Incze, Eva Szentirmay, Eszter Gacs-Baitz, and Csaba Szantay
J. Org. Chem., 2004, ASAP Web Release Date: 03-Aug-2004
DOI:10.1021/jo049209b
Abstract: The first direct synthesis of (+)-lysergic acid (2a) suitable for scale-up has been achieved by the following reaction sequence. Bromoketones 4d or 4g were allowed to react with amine 5 followed by deprotection, and the resulting diketone 6c was transformed into the unsaturated ketone (+-)-7 by the LiBr/Et4N system. Resolution afforded (+)-7, which was further transformed by Schollkopf’s method into the mixture of esters 2e and 2f. Upon hydrolysis the latter mixture afforded (+)-2a. The peptide part of R-ergocryptine (1) was prepared according to the Sandoz method; the stereoefficiency, however, has been significantly improved by applying a new resolution method and recycling the undesired enantiomer. Coupling the peptide part with lysergic acid afforded 1. Having synthetic (+)-7 in hand, we can claim the total synthesis of all the alkaloids which were prepared earlier from (+)-7 that had been obtained through degradation of natural lysergic acid.
The tendency is to push it as far as you can
(Hive Bee)
08-15-04 19:32
No 525546
(Rated as: excellent)
Enantioefficient Synthesis of alpha-Ergocryptine: First Direct Synthesis of (+)-Lysergic Acid
István Moldvai, Eszter Temesvári-Major, Mária Incze, Éva Szentirmay, Eszter Gács-Baitz, and Csaba Szántay
J. Org. Chem., ASAP Article, Web Release Date: August 3, 2004
DOI:10.1021/jo049209
Chemical Research Center, Institute of Biomolecular Chemistry, Hungarian Academy of Sciences, POB 17, H-1525 Budapest, Hungary, and Research Group for Alkaloid Chemistry of the Hungarian Academy of Sciences, Budapest University of Technology and Economics, Gellért tér 4, H-1521 Budapest, Hungary
Received May 11, 2004
Abstract:
The first direct synthesis of (+)-lysergic acid (2a) suitable for scale-up has been achieved by the following reaction sequence. Bromoketones 4d or 4g were allowed to react with amine 5 followed by deprotection, and the resulting diketone 6c was transformed into the unsaturated ketone (±)-7 by the LiBr/Et3N system. Resolution afforded (+)-7, which was further transformed by Schöllkopf's method into the mixture of esters 2e and 2f. Upon hydrolysis the latter mixture afforded (+)-2a. The peptide part of alpha-ergocryptine (1) was prepared according to the Sandoz method; the stereoefficiency, however, has been significantly improved by applying a new resolution method and recycling the undesired enantiomer. Coupling the peptide part with lysergic acid afforded 1. Having synthetic (+)-7 in hand, we can claim the total synthesis of all the alkaloids which were prepared earlier from (+)-7that had been obtained through degradation of natural lysergic acid.
Introduction
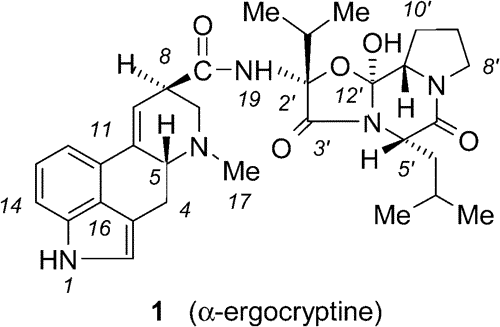
"Ergot alkaloids, of which lysergic acid is representative, are particularly important as they possess the widest spectrum of biological activity found in any family of natural products".1 One of the biologically most important ergot alkaloids is -ergocryptine (1). Its semisynthetic derivative, the so-called bromocryptine, is one of the most widely used drugs in this family (e.g., as a prolactin inhibitor, or an anti-Parkinsonian).2 Great efforts have been devoted to the synthesis of ergot alkaloids during the second half of the last century. Conceptually, retrosynthetic cleavage of the central amide bond devides the problem into two parts, the synthesis of the lysergic acid and of the peptide dilactam moiety.
Scheme 1
The first synthesis of racemic lysergic acid was effected by Woodward and Kornfeld in 1956.3,16 One of their main problems was to prepare ring C from the otherwise easily accessible 3-indolepropionic acid (3), since the ring closure of the corresponding acid chloride occurred at the more reactive pyrrole ring instead of the benzene ring. Thus the Woodward group reduced the pyrrole ring, the amine was protected by benzoylation, and thereafter the ring closure took place regioselevtively as desired. The drawback of this approach is that sooner or later the pyrroline moiety must be reoxidized to a pyrrole ring. It is difficult to perform an enantioefficient synthesis as well, since the method involves introduction of an unnecessary chiral center by the reduction. The earlier described resolution of racemic compound needs further and rather inconvenient steps.4 So far the total synthesis of (±)-2a has been achieved by nine groups, but the number of publications dealing with the synthetic efforts is much higher. Among these approaches one can find about a dozen methods trying to construct the ergoline ring, some of which were successful; others remained at the level of attempt.5 Seven of the nine successful syntheses used the reduced indoline derivative as the starting compound. Oppolzer et al.3 performed the first total synthesis avoiding the reduction step, but their procedure again cannot be scaled up. A second approach to the racemic acid was published recently.6
We decided to construct the ergoline skeleton starting from indole, thus avoiding the reoxidation problem, and at the same time making an enantioefficient synthesis possible.7
An ideal starting material was the so-called Uhle's ketone (4a) having the intact indole ring, although the original synthesis of 4a is rather tedious. Uhle commenced with acetylation and subsequent bromination, and he claimed that the derived bromo-derivative 4b could be subjected successfully to a substitution reaction with various amines.9 Early in the seventies Bowman and co-workers reinvestigated a few results of Uhle's synthesis and established that one of the key steps, alkylation of several types of amines with 4b, always failed.10 The approach starting from 4a by Stoll used the Stobbe condensation as a key step, but the reaction sequence could not be carried out,11 thus the synthesis of lysergic acid starting from Uhle's ketone remained a challenge.
We decided to construct the ergoline skeleton starting from indole, thus avoiding the reoxidation problem, and at the same time making an enantioefficient synthesis possible.7
An ideal starting material was the so-called Uhle's ketone (4a) having the intact indole ring, although the original synthesis of 4a is rather tedious. Uhle commenced with acetylation and subsequent bromination, and he claimed that the derived bromo-derivative 4b could be subjected successfully to a substitution reaction with various amines.9 Early in the seventies Bowman and co-workers reinvestigated a few results of Uhle's synthesis and established that one of the key steps, alkylation of several types of amines with 4b, always failed.10 The approach starting from 4a by Stoll used the Stobbe condensation as a key step, but the reaction sequence could not be carried out, 11 thus the synthesis of lysergic acid starting from Uhle's ketone remained a challenge.
Results and Discussion
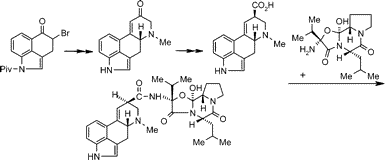
(1) Synthesis of (+)-Lysergic Acid. In 1994 the N-pivaloyl derivative of Uhle's ketone (4c) became easily accessible from 3-indolepropionic acid by Goto's method.12 In connection with our attempt to find a reasonable total synthesis of ergoline skeleton we wished to reinvestigate the cyclization of ring D. We had reported the first successful reaction sequence to this end by applying an unprecedented intramolecular Stobbe condensation taking advantage of a lithium complex formed as an intermediate.13 As a second approach, ring D of the tetracyclic skeleton was formed by an intramolecular Dieckmann condensation of a diester, obtained in a modified Reformatsky reaction of a properly substituted derivative of 4c, followed by elimination of water.14 Neither of these methods, however, could be further elaborated to achieve (+)-lysergic acid.

Scheme 2. Synthesis of 4-Bromo-Uhle's Ketone (4g) from 3a a
Reagents and conditions: (a) (1) powdered KOH + Piv-Cl, CH2Cl2 + THF, (2) SOCl2, (3) AlCl3 + ClCH2COCl, CH2Cl2 (43%, overall);
(b) ref 15 (85%);
(c) HO(CH2)2OH, [I]p-TSA, benzene, reflux, 6 h (81%);
(d) MeNH2, CHCl3, 10-15 °C, 3-4 h (88%);
(e) aq HCl (1 M), acetone, rt, 3 h (97%).
To our pleasant surprise we found that bromoketone 4d, contrary to the literature,10 can be subjected to a substitution reaction with amine 5 providing us with the so far unknown, but much sought-after, even mistakenly claimed9 product (6a), if one has the patience to allow the reaction to proceed at ambient temperature in toluene. The amine component (5) was already known and could easily be prepared.16 After a simple deacylation with methylamine and subsequent deprotection of the ketone the desired compound 6c was for the first time in our hands. The yield was even better if we allowed amine 5 to react with the N-unprotected bromoketone 4g, which had been prepared via ketalization of 4d, N-deacetylation, and a deketalization step in high yields. This sequence yielding 6b and leading from here to6c proved to be a real shortcut.

Scheme 3. Synthesis of Tetracyclic Ketone [(+)-7]a a
Reagents and conditions: (a) 5 + 4d, toluene, 48 h, rt (35%);
(b) 5 + 4g, THF, 24 h, rt (56%);
(c) MeNH2, benzene, 10-15 °C, 1 h (80%);
(d) aq HCl (6 M), 35-40 °C, 1 h then
(e) 6c in CHCl3, LiBr + TEA, 0-5 °C, 12 h (60%, 2 steps);
(f) (-)-dibenzoyl-L-tartaric acid, CH3CN + H2O (1:1) (38%).
The ring closure of 6c leading to the unsaturated ketone 717 by intramolecular aldol condensation seems to be an easy task, but with a great number of well-established agents (from potassium tert-butoxide through super bases to LHMDS) not even a trace of the desired teracyclic compound could be detected. It is worth noting, and not easy to explain, that in the dihydro-indole series this ring closure had been carried out;16 however, similar intramolecular ring closure of compounds with a sulfone group instead of indole nitrogen also failed. An analogous compound of 6c having an indole N-tosyl group and an acetyl group on the second nitrogen in place of the methyl group could be closed by KF, but simultaneously isomerization into a naphthalene derivative also occurred.18 We became successful in performing the reaction by using a LiBr + triethylamine system, which was first used for condensation by Eschenmoser in the case of a different, sulfur-containing compound.19 LiBr or triethylamine alone were totally ineffective. Likely the LiBr leads to a complementary activation of the two carbonyl groups in the presence of basic amine, since lithium ions have a higher affinity toward oxygen than nitrogen. The function of the amine is purely to abstract the proton in the alpha-position with respect to the O-complexed ketone carbonyl. An especially good result was achieved by performing the two consecutive steps (deprotection and ring closure; 6b --> 6c --> 7) without isolation of the intermediate 6c to give a 60% combined yield of crystalline unsaturated ketone 7.
The resolution of 7 was performed with dibenzoyl-tartaric acid. At the same time the optically active ketone was also prepared by degradation of natural lysergic acid,20 and by comparison the absolute configuration of our synthetic compounds was established.

Scheme 4. Synthesis of (+)-Lysergic Acid (2a) from (+)-7a a[/I]
Reagents and conditions: (a) (+)-7 + 8, t-BuOK, THF + t-BuOH, 0 °C, 20 min then + H2O, -5 °C (77%);
(b) aq HCl (2 M), reflux, 30 min (13%);
(c) NaOMe, MeOH, 70-75 °C, 30 min (70%);
(d) HCl/MeOH (6.7 M), 75-80 °C, 45 min (72%);
(e) aq NaOH (5 M), MeOH, 70-80 °C, 2.5 h then aq HCl (6 M) to pH 6.5 (54%).
To proceed, we allowed the optically active 7 to react with the isonitrile derivative 8 in the presence of base21 to yield the formamide derivative 9, followed by acidic hydrolysis. A mixture of lysergic acid (2a) and its epimer (2b) was obtained; after treatment with base almost pure (+)-lysergic acid was isolated as a result of epimerization, although in poor yield.
A much better result was achieved by treating intermediate 9 with base affording a mixture of nitriles (2c:2d, 1:1, 70%) and converting the mixture by Pinner reaction into lysergic acid ester diastereomers (a 3:2 mixture of 2e:2f, 72%). There is no need to separate the two nitriles or esters, since the basic hydrolysis of the mixture of 2e:2f results in pure (+)-lysergic acid (2a) through concurrent hydrolysis and epimerization.22
(2) Improving the Efficiency of the Peptide Part Synthesis. Above we described the synthesis of the (+)-lysergic acid component of alpha-ergocryptine (1). The synthesis of the peptide part has already been described23 by a research group from the Sandoz Pharmaceutical Co. Our task was to improve the efficiency, especially the stereoefficiency of the reaction sequence, and to make a scale-up procedure possible.
At the outset isopropyl malonic ester was oxidized by benzoyl peroxide. According to the original procedure the excess of the benzoyl peroxide was to be eliminated by charcoal, but following this route we observed explosions in about 20% of the cases. To avoid this danger, Na2S2O3 or NaHSO3 was successfully used instead of charcoal. The resulting compound was debenzoylated, and the hydroxyl group was protected as the benzyl ether. Partial hydrolysis of diester 10 gave rise to the half ester (±)-11. In Sandoz's original reaction sequence this acid was resolved by the consecutive application of (-)- and (+)-pseudoephedrine, which process proved to be rather inconvenient and the yield low. Instead of pseudoephedrines we used (+)-1S,2S-2-amino-1-(4-nitrophenyl)propan-1,3-dio
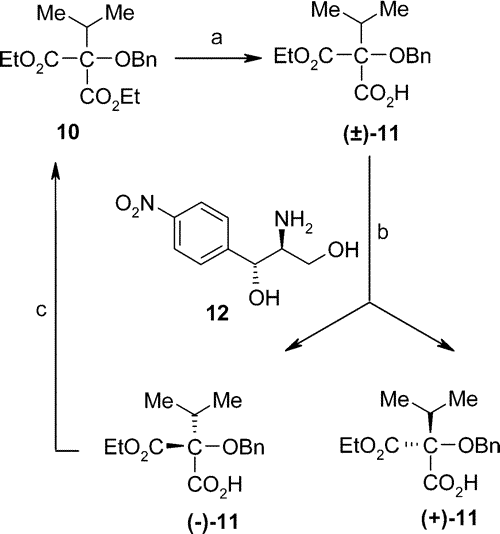
Scheme 5. Modified Resolution of (±)-11a a
Reagents and conditions: (a) ref 23;
(b) (±)-11 + 12, EtOH, rt, 12 h [(+)-11, 38%; (-)-11, 39%;
(c) (EtO)2SO2, acetone, refl., 3 h (90%).
To make the process even more economic, the unwanted S-enantiomer [(-)-11] was esterified with diethyl sulfate to 10. Through this procedure we obtained the original, achiral diester, which we can recycle into the reaction sequence. We may call this manipulation dechiralization.
The so-called aminocyclol hydrochloride (14), the partner needed for coupling with (+)-lysergic acid, was prepared from Z-protected proline. The proline derivative was treated with L-leucine methyl ester p-tolyl sulfonate salt using the mixed anhydride (chloroformic acid ester) method. After deprotection by hydrogenolysis followed by heating, the L-prolyl-L-leucyl lactam (13) was isolated in good yield.
The malonic acid derivative [(+)-11] was transformed to the acid chloride and allowed to react with lactam 13, then deprotected by hydrogenolysis, and the resulting cyclolester hydrolyzed to the so-called cyclolcarboxylic acid. After several steps 14 was btained.
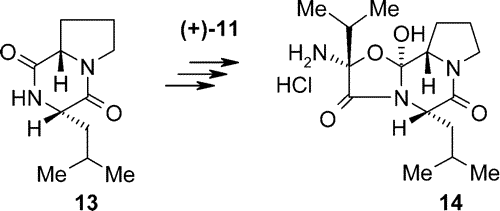
Scheme 6
Several methods were tried for coupling lysergic acid (2a) with the peptide part (14). The most practical route was found by using lysergic acid trifluoroacetate, which was allowed to react with PCl5. The reaction conditions (temperature, the excess of reagent) are critical. The approximate amount (80%) of acyl chloride in the obtained reaction mixture was estimated by IR spectra. By reacting the suspension of the aminocyclol hydrochloride in methylene chloride with lysergic acid chloride hydrochloride25 at -12 °C in the presence of pyridine, alpha-ergocryptine (1) was isolated in 41% yield (as its phosphate salt).26 In addition to its diastereomer alpha-ergocryptinine (15) was obtained (31%) after chromatographic workup.23
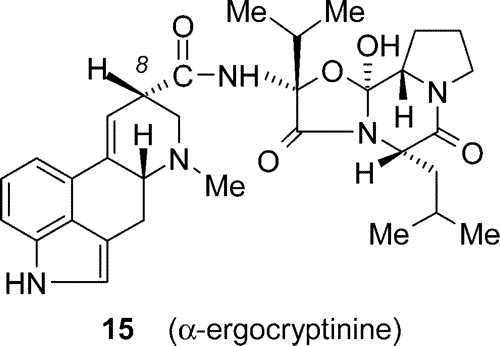
Scheme 7
Since a thermodynamic equilibrium exists between the two stereoisomers in favor of 1 to 15 (3:1) in boiling methanol or in other solvents, in principle there is a possibility to transform 15 into 1 in preparative scale. This aspect, however, was not closely investigated.
The tendency is to push it as far as you can
(Hive Bee)
08-15-04 19:38
No 525548
(Rated as: excellent)
Conclusion
We have shown that a practical, direct synthesis of (+)-lysergic acid is possible while maintaining the indole ring intact throughout the synthesis, thus avoiding the necessity of introducing a needless chiral center by reduction. Furthermore we could perform the resolution of an earlier intermediate, thus avoiding the rather tedious and uneconomic resolution of the end product. Since several natural alkaloids [(+)-isosetoclavine,20a (+)-lysergene,28 (-)-agroclavine28] have been synthesized via a semisynthesis from (+)-7 obtained by degradation of natural (+)-lysergic acid,20 the above approaches from now on can be regarded as the total syntheses of said alkaloids.
By using our modified approach, the peptide part of the alkaloid was synthesized without any side product having the undesired enantiomeric structure, i.e., (-)-11, since the latter was successfully recycled.
Upon coupling the two parts, we have completed an efficient total synthesis of alpha-ergocryptine and alpha-ergocryptinine.
Experimental Section
N-Piv-Uhle's ketone ([b]4c; modified procedure).[/B] To a cold (0-5 °C) solution of 3 (25.0 g, 132.0 mmol) in a mixture of dry CH2Cl2 (1.32 L) and THF (63 mL) were added tetrabutylammonium hydrogen sulfate (5.25 g, 15.4 mmol) and powdered KOH (25.0 g, 446.2 mmol). After the mixture was stirred for 15 min at the above temperature, pivaloyl chloride (67.5 mL, 0.54 mol) in CH2Cl2 (125 mL) was added dropwise. The mixture was stirred for 2 h at room temperature, then cooled again and poured into a mixture of cold water (0.8 L), aq HCl solution (1 M, 75 mL), and CHCl3 (625 mL). The aqueous phase was extracted with CHCl3 (0.5 L), and the combined organic phase was washed with cold water (2 x 0.75 L) and dried. The solvent was evaporated and the residue purified by vacuum distillation (100 °C, 1 mmHg) to remove excess reagent. The obtained crude product (28 g) was dissolved in thionyl chloride (37.0 mL, 309 mmol) with stirring for 15 min at room temperature, then evaporated under reduced pressure. The residue was dissolved in dry CH2Cl2 (260 mL) and dropped (during 0.5 h) into a mixture of aluminum chloride (36.0 g, 270 mmol) and chloroacetyl chloride (32.0 mL, 0.4 mol) in dry dichloromethane (300 mL) at 0-5 °C. The mixture was stirred for 2 h at room temperature, then poured into a mixture of crushed ice (0.5 kg), CHCl3 (1.5 L), and brine (1 L). After extraction, the organic phase was washed with brine (2 x 0.75 L) and cold water (0.75 L) and dried. The crude product was crystallized from ethanol (150 mL) to yield 4c (14.5 g, 43%), in full agreement with the reported data.8a Mp: 167-168 °C.
N-Piv-4-bromo-Uhle's ketone (4d). Preparation of 4d was described in our earlier publication.15
N-Piv-4-bromo-Uhle's Ketone Ethylene Ketal (4e). Protection of bromoketone 4d was carried out by applying a commonly used method (4d, 30.0 g, 89.7 mmol; ethylene glycol, 65 mL; p-TSA, 2.7 g; reflux in 0.6 L of benzene, 6 h, water separating device). After cooling, the mixture was diluted with EtOAc (2.5 L), crushed ice (1.5 L), and aq NH4OH solution (25%, 110 mL). The organic phase was washed with water (2 x 0.6 L) and dried. The crude product was crystallized from ether (100 mL) to afford 27.4 g (81%) of ketal 4e as a cream-colored solid. Mp: 153-155 °C.
4-Bromo-Uhle's Ketone Ethylene Ketal (4f). Methylamine gas was introduced into a solution of 4e (18.9 g, 49.9 mmol) in CHCl3 (300 mL) at 10-15 °C for about 3-4 h. The mixture was washed with water (100 mL) and brine (100 mL) and dried. The crude product was crystallized from diethyl ether (50 mL) to yield 13.04 g (88%) of 4f as a cream-colored solid. Mp: 120-122 °C.
4-Bromo-3,4-dihydro-1H-benzo[c,d]indol-5-one (4g; 4-Bromo-Uhle's Ketone). To a solution of 4f (22.0 g, 74.8 mmol) in acetone (610 mL) at 10-15 °C was added aq HCl solution (1 M, 110 mL). The mixture was stirred for 3 h while the temperature was allowed to warm to room temperature. The organic solvent was evaporated under reduced pressure (water bath: 30-35 °C). The precipitated product was filtered off and washed with water (2 x 75 mL) and ether (2 x 50 mL) to afford 18.0 g (97%) of 4g as a pale brown solid. (The product proved to be unstable at room temperature but it could be stored at -20 °C for a few months without any decomposition.) Mp: 119-122 °C.
N-Piv-4-[N-methyl-N-acetonyl(2',2'-ethylenedioxy)]amino-3,4-dihydro-1H-benzo[c,d]indol-5-one: Alkylation of 5 with 4d (6a). To a solution of 4d (1.12 g, 3.35 mmol) in dry toluene (35 mL) was added amine 5 (1.1 g, 8.3 mmol) in toluene (3.5 mL) at room temperature and the solution was stirred for 48 h. The precipitate formed was filtered off and washed with toluene and the combined filtrate was evaporated in vacuo. Purification by chromatography (eluent: EtOAc/hexane, 2:1) afforded 6a (0.43 g, 35%) as a colorless oil.
4-[N-Methyl-N-acetonyl(2',2'-ethylenedioxy)]amino-3,4-dihydro-1H-benzo[c,d]indol-5-one (6b). (a) Alkylation of 5 with 4g. To a solution of 4g (7.5 g, 30.0 mmol) in dry THF (130 mL) was added amine 5 (8.6 g, 65.1 mmol) in THF (20.0 mL) at room temperature and the solution was stirred for 24 h. The precipitate formed was filtered off and washed with THF and the solvent was evaporated in vacuo (bath: 30-35 °C). The residue was dissolved in a mixture of EtOAc (450 mL) and cold water (180 mL), and the pH was adjusted to ~3 with aq HCl solution (1 M, 54 mL). The organic phase was extracted with aq HCl solution (0.5 M, 2 x 60 mL) and water (50 mL). The aqueous phase was mixed with CHCl3 (450 mL) and the pH was adjusted to ~8 with aq saturated Na2CO3 solution (~40 mL) while the mixture was cooled with an ice bath. After the phases were separated, the aqueous phase was washed with CHCl3 (2 x 100 mL). The combined organic phase was washed with brine (100 mL) and dried. Evaporation (bath: 30-35 °C) of the solvent provided a crude solid (6.83 g, 76%), which was crystallized from a mixture of EtOAc/hexane (1:1, 50 mL) to afford 4.727 g (52.4%) of 6b as a cream-colored solid. A further crop of 6b (0.295 g, 3.3%) was obtained by chromatography of the mother liquor (eluent: EtOAc/hexane, 1:1). Mp: 120-124 °C.
(b) N-Deacetylation of 6a. Methylamine gas was introduced into a solution of 6a (0.5 g, 1.3 mmol) in benzene (50 mL) at 10-15 °C for about 1 h. The mixture was washed with water and brine and dried. The isolation of 6b (0.312 g, 80%) was carried out as described above.
(±)-9,10-Didehydro-6-methylergoline-8-one (7) via Diketone 6c. In the first step 6b (1.6 g, 5.32 mmol) was dissolved in aq HCl solution (6 M, 100 mL) and stirred at 37 °C for 1 h, then cooled in an ice bath. The mixture was mixed with CHCl3 (0.5 L), and the pH was adjusted to ~7 with aq NaOH solution (5 M). After the phases were separated, the aqueous phase was washed with CHCl3 (2 x 100 mL). The combined organic phase was washed with brine (100 mL) and dried. An aliquot part was evaporated (bath: 25-30 °C) in vacuo and the residue was crystallized (ether/hexane, 1:1) to yield 6c as a pale brown solid. Mp: 100-105 °C dec.
As the second step, to a solution of LiBr (2.82 g, 32.5 mmml) in THF (40 mL) at 0-5 °C were added the solution of 6c in CHCl3, obtained after extraction and evaporation to about 100 mL, and TEA (2.82 g, 28 mmol) at 0-5 °C. The mixture was stirred at the above temperature for 12 h, then evaporated (bath: 30 °C). The residue was treated with n-hexane to remove TEA. The obtained oil was purified by chromatography (eluent: CHCl3/MeOH, 10:1) to afford a semisolid product, which was crystallized (EtOAc/hexane, 1:1, 20 mL) to yield 0.758 g (60%) of 729 as pale yellow crystals. Mp: 153-155 °C.
Resolution of (±)-7. To a solution of (±)-7 (595 mg, 2.5 mmol) in a mixture of acetonitrile and water (1:1, 25 mL) at 60 °C was added (-)-dibenzoyl-L-tartaric acid (895 mg, 2.5 mmol) in the same mixture of solvents (12.5 mL). The mixture was stirred for 10-15 min at the above temperature, then cooled to room temperature while being stirred for about an additional 0.5 h. The mixture was kept in a refrigerator overnight. The precipitated crystals were filtered off and washed with the above solvent mixture (5 mL) to yield 585 mg (79%) of salt. This salt (515 mg, 0.864 mmol) was suspended in a mixture of CHCl3 (200 mL) and water (30 mL) at 0-5 °C and the pH was adjusted to ~9 with aq NaOH solution (1 M, 2 mL). After the phases were separated, the aqueous phase was washed with CHCl3 (2 x 50 mL). The combined organic phases were washed with water, dried, and evaporated. The residue was crystallized (hexane/ether, 1:1, 10 mL) to yield (+)-7 (226 mg, 38%) as a yellow crystal. Mp: 165-169 °C.
Isolation of (-)-7. The mother liquor of the first crystallization was evaporated in vacuo until an aqueous solution was obtained. The solution was diluted with water (50 mL) and CHCl3 (300 mL) and cooled at 0-5 °C. The pH of the mixture was adjusted to ~9 with aq NaOH solution (1 M, 3 mL). After the phases were separated, the organic phase was washed with water, dried, and evaporated. The residue was crystallized (hexane/ether, 1:1, 10 mL) to yield crude (-)-7 (306 mg) as a solid. This product (299 mg, 1.256 mmol) was resolved with (+)-dibenzoyl-D-tartaric acid as described above to yield 262 mg of salt (35%). The salt was treated with aq NaOH in CHCl3 as desribed above and the crude product was crystallized to afford 118 mg (20%) of (-)-7. Mp: 165-168 °C.
Preparation of (+)- E/Z-Formamide 9. To a solution of (+)-7 (453 mg, 1.9 mmol) in dry THF (10 mL) at room temperature were added p-toluenesulfonylmethyl isocyanide (374 mg, 1.9 mmol) then t-BuOK (426 mg, 3.8 mmol) in a mixture of THF (5 mL) and t-BuOH (2 mL) at 0 °C. After being stirred for 20 min, the mixture was cooled to -5 °C, diluted with water (50 mL), and extracted with CHCl3 (3 x 50 mL). The combined organic phases were washed with brine and dried. The crude product was crystallized (hexane/ether, 1:1, 10 mL) to afford 637 mg (77%) of 9 as a cream-colored solid. Mp: 178-184 °C dec.
(+)-8-Cyano-9,10-didehydro-6-methylergol
For structure determination of the crude product, an aliquot part was purified by column chromatography (eluent: CHCl3/acetone, 10:1) to afford pure 8 beta-isomer31 2c. Mp: 140-160 °C dec.
The chemical shifts of the 8alpha-isomer32 2d were determined in the mixture.
Preparation of Esters (2e:2f). Isomer mixture 2c:2d (300 mg, 1.2 mmol) was dissolved in HCl/MeOH (6.7 M, 30 mL) and the solution was refluxed for 45 min. After being cooled with an ice bath, the mixture was poured into a mixture of CHCl3 (100 mL) and crushed ice (150 g). The pH was adjusted to ~7-8 with aq saturated Na2CO3 solution (120 mL). After the phases were separated, the aqueous phase was washed with CHCl3 (20 mL). The combined organic phase was washed with water, dried, and evaporated to yield 246 mg (72%) of a mixture of isomers (2e:2f) as a semisolid material, which was subjected to further transformations. Mp: 131-139 °C.
For structure determination of the crude product, an aliquot part was purified by crystallization from benzene to afford pure (+)-lysergic acid methyl ester33 2e as a cream-colored solid, in agreement with the reported data.6,34 Mp: 164-166 °C.
The chemical shifts of the 8alpha-isomer35 2f were determined in the mixture.
(+)-Lysergic Acid 2a. (a) Starting from Esters 2e:2f. Isomer mixture 2e:2f (340 mg, 1.2 mmol) was dissolved in MeOH (30 mL) and an aq NaOH solution (5 M, 7 mL) was added. The mixture was then stirred at 75-80 °C for 45 min. The hot solution was treated with charcoal and filtered. The organic solvent was removed by evaporation, and the aqueous solution was diluted with water (10 mL) and cooled to 0-5 °C. The solution was acidified to pH 6.5 with aq HCl solution (6 M) and stirred for a further 1-2 h at 0-5 °C while a solid was formed. The precipitate was filtered off and washed with cold water (3 x 2 mL) and acetone (3 x 2 mL) to afford 174 mg (54%) of 2a as a pale brown solid, in agreement with the reported data.6,34a Mp: 230-240 °C dec.
(b) Acidic Hydrolysis of (+)-9 and Epimerization. Formamide 9 (0.5 g, 1.1. mmol) was dissolved in aq HCl solution (2 M, 40 mL) and refluxed for 30 min. After cooling, the pH of the solution was adjusted to 6.5 with aq saturated NaHCO3 solution and evaporated in vacuo to dryness. The residue (140 mg, 45%) was purified by chromatography (eluent: CHCl3/MeOH/cm3 NH4OH solution, 5:5:0.1) to afford 50 mg (13%) of 2a:2b. The NMR data of 2a have been described above, and the chemical shifts of 2b were determined from the mixture.
In the next step 100 mg (0.37 mmol) of 2a:2b was dissolved in MeOH (10 mL) at room temperature and KOH (100 mg, 1.8 mmol) was added in a mixture of MeOH (2 mL) and water (1 mL). The mixture was stirred for 48 h, then a further portion of KOH (100 mg in the same solvents) was added, and the mixture was stirred for 48 h. The solution was acidified to pH 6.5 with aq HCl solution (1 M) and evaporated in vacuo to dryness. The residue was purified by chromatography (eluent: CHCl3/MeOH/cm3 NH4OH solution, 5:5:0.1) to afford 50 mg (50%) of 2a.
Resolution of (±)-11. To a solution of (±)-11 (77.2 g, 0.276 mol) in dry EtOH (0.5 L) was added 12 (58.45 g, 0.276 mol) at room temperature. The mixture was stirred for a few minutes, then kept at room temperature overnight. The precipitated crystals were filtered off, washed with cold EtOH, and recrystallized from EtOAc (1.8 L) to afford 54 g (80%) of salt. Mp: 145-147 °C. To a suspension of the above salt (54 g) in CH2Cl2 (240 mL) were added crushed ice (100 g) and phosphoric acid (85%, 19.4 mL). After the solution was stirred for a few minutes, the phases were separated. The aqueous phase was extracted with CH2Cl2 (3 x 200 mL). The combined organic phase was washed with aq HCl solution (2 M, 2 x 100 mL) and water and dried. The solvent was removed under reduced pressure to yield 29.2 g (38%) of (+)-isopropylbenzyloxymalonic acid monoethyl ester [(+)-11] as a colorless oil. (Compound 12 could be recovered from the aqueous phase.)
The mother liquor formed after the first crystallization and recrystallization was evaporated in vacuo to dryness. The obtained oily salt (80 g) was treated with phosphoric acid in CH2Cl2 as described above to afford 35.4 g (39%) of (-)-11 as a crude product.
Dechiralisation of (-)-11. To a solution of (-)-11 (35 g, 125 mmol) in dry acetone (0.5 L) were added dry K2CO3 (34.5 g, 0.25 mol) and diethyl sulfate (24.6 mL, 187 mmol). The mixture was heated to reflux for 3 h, then cooled to room temperature and poured into cold water (1 L). The resulting oil was separated and the aqueous phase was extracted with ether (2 x 100 mL). The combined organic phase was washed with aqueous NaHCO3 solution (5%, 2 x 100 mL) and water and dried. The solvent was removed under reduced pressure and the crude product (41.3 g) was purified by distillation (bp: 130-134 °C, 0.5 mmHg) to give 34.8 g (90%) of isopropylbenzyloxymalonic acid diethyl ester (10).
Acknowledgment
The Found OTKA T-046015 is gratefully acknowledged. Special thanks are due to Dr. Ágnes Gömöry for the MS spectra and Ms. Gabriella Hanek and Ms. Mónika Jeruska for technical assistance.
The tendency is to push it as far as you can
(Hive Bee)
08-15-04 20:00
No 525552
(Rated as: excellent)
References
1. Bur, S. C.; Padwa, A. Org. Lett. 2002, 4, 4135-4137.
2. The Merck Index, 12th ed.; Merck and Co. Inc.: Whitehouse Station, New York, 1996; p 231.
3. (a) Somei, M.; Yokoyama, Y.; Murakami, Y.; Ninomiya, I.; Kiguchi, T.; Naito, T. Recent Synthetic Studies on the Ergot Alkaloids and Related Compounds; The Alkaloids, Vol. 54; Cordell, G. A., Ed.; Academic Press: San Diego, CA, 2000; pp 191-257.
(b) Ninomiya, I.; Kiguchi, T. Ergot Alkaloids; The Alkaloids, Vol. 38; Brossi, A., Ed.; Acadenic Press: San Diego, CA, 1990; pp 1-156 and citations therein.
4. Stoll, A.; Hofmann, A. Helv. Chim. Acta 1943, 26, 922-928.
5. E.g.: (a) Marino, J. P., Jr.; Osterhout, M. H.; Padwa, A. J. Org. Chem. 1995, 60, 2704-2713.
(b) Ralbovsky, J. L.; Scola, P. M.; Sugino, E.; Burgos-Garcia, C.; Weinreb, S. M. Heterocycles 1996, 43, 1497-1512.
(c) Sader-Bakaouni, L.; Charton, O.; Kunesch, N.; Tillequin, F. Tetrahedron 1998, 54, 1773-1782.
(d) Liras, A.; Lynch, C. L.; Fryer, A. M.; Vu, B. T.; Martin, S. F. J. Am. Chem. Soc. 2001, 123, 5918-5924.
(e) Lee, K. L.; Goh, J. B.; Martin, S. F. Tetrahedron Lett. 2001, 42, 1635-1638.
6. Hendrickson, J. B.; Wang, J. Org. Lett. 2004, 6, 3-5.
7. It is worth mentioning that one of our abandoned earlier routes was later developed for the synthesis of racemic LSD.8 Galambos, G.; Szántay, Cs., Jr.; Tamás, J.; Szántay, Cs. Heterocycles 1993, 36, 2241-2245.
8. Saá, C.; Crotts, D. D.; Hsu, G.; Vollhardt, K. P. C. Synlett 1994, 487-489.
9. Uhle, F. C. J. Am. Chem. Soc. 1951, 73, 2402-2403.
10. Bowman, R. E.; Evans, D. D.; Guyett, J.; Weale, J.; White, A. C. J. Chem. Soc., Perkin Trans. 1 1973, 760-766.
11. (a) Stoll, A.; Rutschmann, J.; Petrzilka, T. Helv. Chim. Acta 1950, 33, 2257-2261.
(b) Stoll, A.; Rutschmann, J. Helv. Chim. Acta 1952, 35, 141-147.
12. (a) Teranishi, K.; Hayashi, S.; Nakatsuka, S.; Goto T. Tetrahedron Lett. 1994, 35, 8173-8176.
(b) Although Goto's method has given a really short route to tetracyclic 4a and 4c, the application of the expensive and dangerous n-BuLi at -78 °C can interfere with their scale-up preparation. In our modification powdered KOH was applied for deprotonation of 3 at 0 °C. After acylation at room temperature, the obtained crude product was transformed directly to 4c in 43-46% overall yield.
13. (a) Moldvai, I.; Temesvári-Major, E.; Gács-Baitz, E.; Egyed, O.; Gömöry, Á.; Nyulászi, L.; Szántay, Cs. Heterocycles 1999, 51, 2321-2333.
(b) Moldvai, I.; Temesvári-Major E.; Gács-Baitz, E.; Egyed, O.; Nyulászi L.; Szántay, Cs. Hetrocycles 2000, 53, 759.
14. Incze, M.; Moldvai, I.; Temesvári-Major, E.; Dörnyei, G.; Kajtár-Peredy, M.; Szántay, Cs. Tetrahedron 2003, 59, 4281-4286.
15. Moldvai, I.; Temesvári-Major, E.; Balázs, M.; Gács-Baitz, E.; Egyed, O.; Szántay, Cs. J. Chem. Res. (S) 1999, 687; J. Chem. Res. (M) 1999, 3018-3029.
16. Kornfeld, E. C.; Fornefeld, E. J.; Kline, G. B.; Morrison, D. E.; Jones, G.; Woodward, R. B. J. Am. Chem. Soc. 1956, 78, 3087-3114.
17. The first synthesis of racemic 7 has been performed via oxidation of the corresponding alcohol with the 2,3-dihydroindole ring. Only one characterization datum (mp 145-148 °C) of (±)-7 has been described. See: Bach, N. J.; Hall, D. A.; Kornfeld, E. J. J. Med. Chem. 1974, 17, 312-314.
18. (a) Craig, J. C.; Hurt, S. D. J. Org. Chem. 1979, 44, 1113-1117. (b) Moldvai, I.; Gács-Baitz, E.; Temesvári-Major, E.; Incze, M.; Poppe, L.; Szántay, Cs. Heterocycles. In press.
19. (a) Roth, M.; Dubs, P.; Götschi, E.; Eschenmoser, A. Helv. Chim. Acta 1971, 54, 710-734.
(b) Waldvogel, E.; Engeli, P.; Küsters, E. Helv. Chim. Acta 1997, 80, 2084-2099.
20. (a) Bernardi, L.; Gandini, E.; Temperilli, A. Tetrahedron 1974, 30, 3447-3450. A few characterization data of (+)-7 have been described.
(b) Bach, N. J.; Kornfeld, E. C. Tetrahedron Lett. 1974, 3225-3227.
21. (a) Schöllkopf, U.; Schröder, R.; Blume, E. Justus Liebigs Ann. Chem. 1972, 766, 130-141. (b) Schöllkopf, U.; Schröder, R. Angew. Chem. 1973, 85, 402-403.
22. It is worth mentioning that Rapoport and co-workers published a method for synthesizing an advanced intermediate of the Ergot alkaloids, an optically active amino derivative of Uhle's ketone, following an entirely different route. Hurt, C. R.; Lin, R.; Rapoport, H. J. Org. Chem. 1999, 64, 225-233.
23. Stadler, P. A.; Guttmann, S.; Hauth, H.; Huguenin, R. L.; Sandrin, E.; Wersin, G.; Willems, H.; Hofmann, A. Helv. Chim. Acta 1969, 52, 1549-1564.
24. Kollonitsch, J.; Haás, A.; Kraut, M.; Gábor V. Acta Chim. Hung. 1955, 6, 381-395; Chem. Abstr. 1955, 49, 6872h.
25. Magó-Karácsony, E.; Balogh, T.; Borsi, J.; Elek, S.; Polgari, I.; Löwinger, L. Hungarian patent 156.385, 1969; Chem. Abstr. 1970, 72, 32105r.
26. Stoll, A.; Hofmann, A. Helv. Chim. Acta 1943, 26, 1570-1601.
27. Bandula, R.; Vasilesu, M. Rev. Roum. Chim. 1995, 40,1189-1195; Chem. Abstr. 1996, 125, 114925k.
28. Wheeler, W. J. Tetrahedron Lett. 1986, 27, 3469-3470. 29. The formation of ring D was confirmed by the presence of an NOE effect between H-9 and H-12 protons. The chemical shifts of C-9 and C-10 carbons (118.8 and 156.0 ppm, respectively) are also characteristic for an alpha,beta-unsaturated ketone unit.
30. Evidence for the given stereochemistry of the double bond in 9 was provided by NOE experiments. Irradiation of the H-7beta proton (3.37 ppm) leads to observation of an NOE at the formyl proton (8.09 ppm), while the tosyl protons gave NOE with H-9 (7.88 ppm).
31. The beta-orientation of the nitril group at C-8 follows from the vicinal coupling constant (10.4 Hz) between H-8 and H-7beta protons.
32. The configurational change at C-8 in comparison with 2c is confirmed by the coupling constant values of H-8 with H-7alpha and H-7beta (3.0 and 3.4 Hz, respectively).
33. The configuration at C-8 was determined by the NOE effect between H-8 and H-7alpha, and by the value of the coupling constant between H-8 and H-7beta protons (10.5 Hz).
34. (a) Smith, S.; Timmis, G. M. J. Chem Soc. 1936, 1440-1444. (b) Jacobs, W. A.; Craig, L. C. J. Biol. Chem. 1934, 104, 547-549. 35. The epimerization at C-8 is best reflected by the values of the coupling constants between H-8 and H-7alpha and H-7beta protons.
36. In DMSO-d6 solution 2a experienced a rapid C-8 epimerization and as a result 2b appeared in the isomer mixture.
Reference 1
A Novel Sequential Aminodiene Diels-Alder Strategy for the Rapid Construction of Substituted Analogues of Kornfeld’s Ketone
Scott K. Bur and Albert Padwa
Org. Lett., 2002, 4(23), 4135-4137
DOI:10.1021/ol0268992
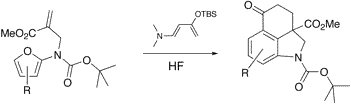
Abstract: Through a novel sequence of aminodiene Diels-Alder reactions, amidofurans 18a-c were converted to tricyclic ketones 21a-c in moderate to good yields. Ketone 21a could be converted to Uhle´’s ketone (6) by cleaving the tert-butyl carbamate and oxidatively removing the methyl ester. Tricycle 21a readily underwent bromination to give 22. Formation of the corresponding enol triflate 25 followed by carbonylation gave ester 27, which was then coupled with N-methyl propriolamide to furnish 26.
Reference 5a
An Approach to Lysergic Acid Utilizing an Intramolecular Isomuenchnone Cycloaddition Pathway
Marino, Joseph P., Jr.; Osterhout, Martin H.; Padwa, Albert
J. Org. Chem., 1995, 60(9), 2704-2713
No DOI found
[upload][/upload]
Reference 5c
Intramolecular Diels-Alder reaction of dinitro-olefin derivatives of furan for the preparation of a versatile tool: 3,7-dinitro-11-oxatricycloundec-9-ene
Sader-Bakaouni, Lina; Charton, Olivier; Kunesch, Nicole; Tillequin, Francois
Tetrahedron, 1998, 54(9), 1773-1782
DOI:10.1016/S0040-4020(97)10411-2
Abstract: Recurrent addns. of nitromethane on furfuraldehyde followed by an intramol. Diels-Alder reaction allowed the preparation of the title compound I in good yield with excellent stereoselectivity. Aromatization, ether cleavage and stereocontrolled oxidation reactions give evidence of the synthetic versatility of this adduct in the preparation of ergot alkaloids and valienamine bicyclic analogs.
Reference 5d
Applications of Vinylogous Mannich Reactions. Total Syntheses of the Ergot Alkaloids Rugulovasines A and B and Setoclavine
J. Am. Chem. Soc., 2001, 123 (25), 5918-5924
Spiros Liras, Christopher L. Lynch, Andrew M. Fryer, Binh T. Vu, and Stephen F. Martin
DOI:10.1021/ja010577w S0002-7863(01)00577-7
Abstract: Concise syntheses of the Ergot alkaloids rugulovasine A (3a), rugulovasine B (3b), and setoclavine (2) have been completed by strategies that feature inter- and intramolecular vinylogous Mannich reactions as the key steps. Thus, the first synthesis of 3a,b commenced with the conversion of the known indole 17 into 24 via the addition of the furan 22 to the iminium ion 21, which was generated in situ from the aldehyde 19. Cyclization of 24 by a novel SRN1 reaction followed by removal of the N-benzyl group furnished a mixture (1:2) of 3a and 3b. In an alternative approach to these alkaloids, the biaryl 35 was reduced with DIBAL-H to give an intermediate imine that underwent spontaneous cyclization via an intramolecular vinylogous Mannich addition to provide 36a,b. N-Methylation of the derived benzyl carbamates 37a,b followed by global deprotection gave a mixture (2:1) of rugulovasines A and B (3a,b). Setoclavine (2) was then prepared from the biaryl 41 using a closely related intramolecular vinylogous Mannich reaction to furnish the spirocyclic lactones 42a,b. These lactones were subsequently transformed by hydride reduction and reductive methylation into the ergoline derivatives 43a,b, which were in turn converted into 2 by deprotection and solvolytic 1,3-rearrangement of the allylic hydroxyl group.
Reference 5e
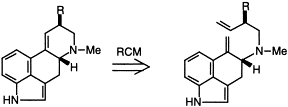
Novel entry to the Ergot alkaloids via ring closing metathesis
Katherine L. Lee, Jane Betty Goh and Stephen F. Martin
Tet. Lett., 2001, 42(9), 1635-1638
DOI:10.1016/S0040-4039(01)00002-8
Reference 6
Post 476514 (Lego: "A New Synthesis of Lysergic Acid", Tryptamine Chemistry)
Post 480281 (Rhodium: "Some syntheses of LSD and analogs", Tryptamine Chemistry)
Reference 8
A cobalt-catalyzed entry into the ergot alkaloids: Total synthesis of (±)-Lysergene and (±)-LSD
Saá, C.; Crotts, D. D.; Hsu, G.; Vollhardt, K. P. C.
Synlett, 1994, 487-489
No DOI found
../rhodium/pdf /lsd.cobalt.pd
Abstract: Cocyclization of 4-ethynyl-3-indoleacetonitriles with alkynes in the presence of CpCO(CO)2 gives rise to the ergoline derivatives 6-9, two of which were transformed into racemic lysergence and LSD, respectively.
Reference 9
Amino derivatives of 5-keto-1,3,4,5-tetrahydrobenz[cd]indole
Uhle, F. C.
J. Am. Chem. Soc., 1951, 73, 2402-2403.pdf
No DOI found
Abstract: 1-Acetyl-5-keto-1,3,4,5-tetrahydrobenz[c
Reference 11
Post 410147 (bottleneck: "Here are a few old review articles: ...", Tryptamine Chemistry)
Reference 12a
Post 400179 (bottleneck: "Even though people don't appear too interested", Tryptamine Chemistry)

Reference 14
Chemistry of indoles carrying a basic function. Part 8: A new approach to the ergoline skeleton
Mária Incze, István Moldvai, Eszter Temesvári-Major, Gábor Dörnyei, Mária Kajtár-Peredy and Csaba Szántay
Tetrahedron , 2003, 28(9), 4281-4286
DOI:10.1016/S0040-4020(03)00630-6
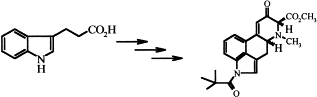
Reference 15
Chemistry of Indoles carrying a Basic Function. Part 5. Some Observations while Constructing an Ergoline Ring by Stobbe Reaction
Moldvai, I.; Temesvári-Major, E.; Balázs, M.; Gács-Baitz, E.; Egyed, O.; Szántay, Cs.
J. Chem. Res. (S), 1999, 687
No DOI found
Abstract: Tricyclic indole derivative 11 affords the imide 12 under Stobbe reaction conditions rather than the expected
intermediate containing an ergoline ring.
J. Chem. Res. (M), 1999, 3018-3029
No DOI found
Reference 16
Total synthesis of lysergic acid
Kornfeld, Edmund C.; Fornefeld, E. J.; Kline, G. Bruce; Mann, Marjorie J.; Morrison, Dwight E.; Jones, Reuben G.; Woodward, R. B.
J. Am. Chem. Soc., 1956, 78, 3087-3114
No DOI found
[upload][/upload]
Abstract:
Reference 17
Descarboxylysergic acid (9,10-didehydro-6-methylergoline)
Bach, Nicholas J.; Hall, David A.; Kornfeld, Edmund C.
J. Med. Chem., 1974), 17(3), 312-314
No DOI found
[upload][/upload]
Abstract: (+-)-Descarboxylysergic acid maleate (I) [51921-12-1] was prepared by electrolytic reduction of 8b-acetoxy-1-acetyl-9,10-didehydro-2,3- dihydro-6-methylergoline [51867-09-5] to 1-acetyl-9,10-didehydro-2,3-dihydro -6-methylergoline maleate [51867-41-5], followed by hydrolysis and MnO2 oxidation I gave pharmacol. responses similar to other ergot derivs., but was usually less active than the best standard drugs. Activity in relation to substitution and steric factors is discussed.
Reference 18a
Craig, J. C.; Hurt, S. D.
J. Org. Chem., 1979, 44, 1113-1117
DOI:
[upload][/upload]
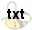
Abstract:
Reference 18b
Chemistry of Indoles Carrying a Basic Function. Part IX. Unexpected Cyclizations of Diketones Derived from Uhle‘s Ketone
István Moldvai, Eszter Gács-Baitz, Eszter Temesvári-Major, Mária Incze, László Poppe, and Csaba Szántay
Heterocycles, 2004, 64,
No DOI found

Reference 22
Enantiospecific Synthesis of (R)-4-Amino-5-oxo-1,3,4,5-tetrahydrobenz
Clarence R. Hurt, Ronghui Lin, and Henry Rapoport
J. Org. Chem., 1999, 64 (1), 225 -233
DOI:10.1021/jo981723s
Abstract: We report a new strategy for the enantiospecific synthesis of (R)-4-amino-5-oxo-1,3,4,5-tetrahydrobenz
corresponding beta-amino alcohol.
Reference 34
../rhodium /ergotin
The tendency is to push it as far as you can