Summary
The syntheses of high specific activity 125I- and 123I-labelled (R)- and (S)-2,5-dimethoxy- 4-iodophenylisopropylamine (DOI) are described. Three radiosynthetic routes, two of which involved use of amine protecting groups and one of which utilized the free base, were compared to maximize the radiochemical yields and specific activities of the products. The method which provided the highest yields utilized the free amine with no protecting group in aqueous acidic solvent with chloramine-T oxidant. Final radiochemical yields of ca. 80% were achieved for both 125I and 123I incorporation. The specific activities of the 125I-labelled products averaged 1100 Ci/mmol and the 123I-labelled products were >20,000 Ci/mmol.
Introduction
Extensive structure-activity studies1 have demonstrated that the most potent hallucinogens are phenethylamines that possess a 2,4,5-trisubstituted aromatic nucleus and an alpha-methyl substituent attached to the side chain (amphetamine derivatives). Among the most active of these are 1-(2,5-dimethoxyphenyl)-2-aminopropanes substituted at the 4-position with a methyl group (DOM), a bromine (DOB), or an iodine (DOI). These compounds are two orders of magnitude more potent than mescaline as psychoactive agents in humans, and animal model studies corroborate their high potency2.
The development of an asymmetric synthesis for these types of compounds3 has allowed the preparation of optically active isomers. The affinities for the enantiomers of DOM, DOB, and DOI have been measured at both 5-HT1 and 5-HT2 serotonin sites4. These compounds have shown good selectivity for the 5-HT2 site, with the R-isomers possessing greater potency than the S-isomers in both animals and man. Recently (±)-[3H]-DOB has been investigated for labelling rat cortical 5-HT2 receptors5,6, and has high affinity for cortical binding sites. The maximum specific activity of the mono-tritiated ligand (29 Ci/mmol) limits its application, and the use of a racemic ligand mixture complicates the interpretation of the data, since it is a mixture of two pharmacologically distinct compounds. We report here the preparation of R-(-)-DOI and S-(+)-DOI, labelled separately with 125I and 123I, in high specific activity. These have proven effective both as radioligands in receptor site studies7 and in autoradiographic studies of receptor localization8.
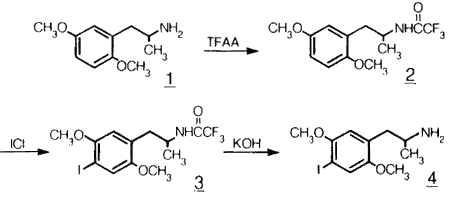
Scheme 1.
Synthesis of the N-trifluoroacetyl protected precursor (2) and cold DOI.
Previous work with these compounds has included radioiodination of the phthalimide protected 2,5-dimethoxyamphetamine precursor with low specific activity 131I and 123I9,10. Glennon and co-workers have recently reported a radiosynthesis of racemic 125I-DOI using an N-trifluoroacetyl protected triazene precursor which provides a radiochemical yield of ca. 3%11. Synthesis of high specific activity racemic 77Br-DOB utilizing the N-trifluoroacetyl protecting group, generated in situ, has also been reported12. The preparation and chemical identification of the N-trifluoroacetyl protected enantiomeric bases (R)-2 and (S)-2 (Scheme 1) are reported here. The enantiomers of 1-(2,5-dimethoxyphenyl)-2-aminopropane were prepared in three steps from 1-(2,5-dimethoxyphenyl)-2-propanone in about 55% overall yield3. Cold aromatic iodination of the amides was effected with ICl in acetic acid9 in 80-85% yield.
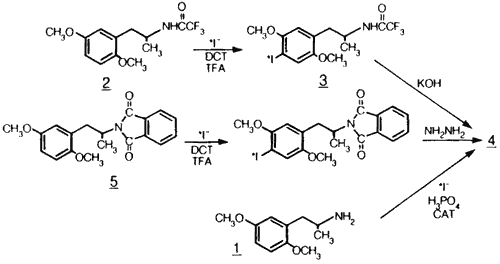
Scheme 2.
Three radiosynthetic routes to radioiodinated DOI.
In a previous report13, the synthesis of cold (R)-DOI afforded a poor yield in the iodination step, and in addition used a procedure not adaptable to radiochemical iodination. Unlabelled (S)-DOI has been previously obtained4 by chemical resolution of racemic DOI by multiple crystallizations of the salt with (+)-tartaric acid, a procedure which is likewise unsuitable for preparation of a radiolabelled form of DOI.
Three radiosynthetic routes for the preparation of 125I-DOI and 123I-DOI are reported here (Scheme 2). Two of the routes involved the use of protecting groups, radioiodination and subsequent deprotection; the third utilized the free base.
Experimental
Materials and Methods
Melting points were taken on a Mel-Temp apparatus and are uncorrected. 1H-NMR spectra were recorded on a Chemagnetics A-200 MHz spectrometer at Purdue University or on the 200-MHz UC Berkeley Chemistry Department NMR. Chemical shifts are reported in delta values (parts per million) relative to an internal reference of Me4Si. Abbreviations used in the NMR analysis include the following: bs, broad singlet; d, doublet; dd, doublet of doublets; m, multiplet; p, pentet; and s, singlet. Only one NMR analysis is reported for each pair of enantiomers, as the spectra of optical antipodes were virtually identical. Mass spectral analysis was performed on a Finnegan 2000 spectrometer. Optical rotations were recorded using a Perkin-Elmer 241 polarimeter. Microanalyses were performed at the Purdue Microanalysis Laboratory or at the University of California, Berkeley, Chemistry Department Microanalysis Laboratory, and all values were within 0.4% of the calculated composition.
High concentration, no-carrier-added (purportedly 17 Ci/mg iodide) sodium 125I-iodide in 0.1M NaOH was purchased from New England Nuclear Corporation. No-carrier-added sodium 123I-iodide in dilute NaOH was purchased from AECL, Canada and Crocker Nuclear Laboratory, University of California, Davis. High performance liquid chromatography (HPLC) was performed with a Waters Associates 590 pump and U6K injector with a Waters Model 450 UV detector (254 nm) and a NaI(Tl) detector in series for absorbance and radioactivity measurements. Two HPLC systems were utilized and kept completely separate so that high specific activity products could be collected on one of the two. Quantitation of the UV and radioactivity peaks was accomplished with a Spectra-Physics Model 4270 integrator. HPLC separations were carried out using Waters C18 reverse phase columns eluted with methanol/water mixtures buffered with 0.2% triethylamine and conc. phosphoric acid to bring the pH to 7.6.
Cold Chemical Syntheses
(R)-1-(2,5-dimethoxyphenyl)-2-trifluoroacetamidopropane ((R)-2)
(R)-2,5-dimethoxyamphetamine ((R)-1) (2.00g, 10.24 mmol) prepared by the method of Nichols et al.3 was stirred into dry benzene (200 mL) under a nitrogen atmosphere. Trifluoroacetic anhydride (10.75g, 51.20 mmol) was then added all at once and the reaction was stirred at room temperature for 30 min. After reflux for an additional 30 min, the solvent was removed in vacuo and the resulting solid was crystallized from ethyl acetate/hexanes to yield feathery white crystals: 2.75g (92.3%); mp 120°C; [α]D +12.27° (c, 1, CHCl3).
(S)-1-(2,5-dimethoxyphenyl)-2-trifluoroacetamidopropane ((S)-2)
(S)-2,5-dimethoxyamphetamine (S-1) treated in an identical manner produced feathery white crystals: 2.67g (89.6%); mp 120°C; [α]D -11.65° (c, 1, CHCl3).
(R)-1-(2,5-dimethoxy-4-iodophenyl)-2-trifluoroacetamidopropane [(R)-3]
Iodine monochloride (0.290g, 1.716 mmol) was added to 5 mL of glacial acetic acid with stirring under an argon atmosphere. R-2 (0.500g, 1.716 mmol) was transferred to the flask in 5 mL of hot glacial acetic acid, and the reaction was stirred at room temperature for 1 h. After heating at 60°C for 1 h, the mixture was cooled, flooded with H2O (75 mL), extracted with CHCl3 (3 x 25 mL), and the pooled extracts were washed with 5% NaHCO3 (50 mL), and H2O (50 mL). The organic extract was dried (Na2SO4) and the solvent was removed by rotary evaporation. The resulting solid was crystallized from ethyl acetate, affording a fine white crystalline product: 0.610g (85.2%); mp 192-193°C; [α]D +23.15° (c, 1, CHCl3).
(S)-1-(2,5-dimethoxy-4-iodophenyl)-2-trifluoroacetamidopropane ((S)-3)
(S)-2 treated in an identical manner afforded white crystalline product: 0.576g (80.4%); mp 191-192°C; [α]D -22.90° (c, 1, CHCl3).
(R)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane ((R)-4)
To (R)-3 (0.200g, 0.479 mmol) was added 20 mL of 2-propanol and 1 mL of aqueous 2N KOH under an argon atmosphere. This was allowed to stir at room temperature for 4 h, and the solvent was then removed by rotary evaporation. The resultant slurry was dissolved in 3N NaOH (25 mL) and extracted with CHCl3 (3 x 25 mL). The CHCl3 extracts were pooled and the amine was extracted with 3N HCl (2 x 25 mL). The pooled acidic aqueous extract was then made strongly basic by addition of 5N NaOH and was re-extracted with CHCl3 (3x25 mL). The pooled organic extracts were dried (Na2SO4) and the solvent was removed by rotary evaporation. Drying under high vacuum afforded a white solid: 0.149g (96.7%); mp 96-97°C. This free base (0.132g, 0.411 mmol) was then dissolved in absolute ethanol and acidified with conc. HCl. The solvent was removed in vacuo and the residual solid was crystallized from 2-propanol/diethyl ether to yield a white crystalline hydrochloride salt: 0.121g (82.3%); mp 232°C; [α]D -11.23° (c, 1, H2O) (lit.13 mp 218-219°C; [α] -12.0°).
(S)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane ((S)-4)
An exact replication of the above method using (S)-3 afforded the white solid free base: 0.149g (96.7%), mp 96-97°C, and a white crystalline hydrochloride salt: 0.120g (80.5% from the base), mp 232°C; [α]D +11.47° (c, 1, H2O), (lit.4 mp 224-225°C; [α] +12.6°).
(±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
(Racemic 4 via Chloramine-T oxidant with iodide as the limiting reagent):
To racemic 2,5-dimethoxyamphetamine9 (195 mg, 1.0 mmol) in 30 mL of 2.0M H3PO4 was added KI (33 mg, 0.20 mmol) and chloramine-T (100 mg, 0.44 mmol). The reaction was stirred at room temperature for 25 min and quenched with sodium metabisulfite. The mixture was made basic with conc. NaOH and extracted with CH2Cl2 (3x25 mL), and the solvent was removed by rotary evaporation. Racemic 4 was separated from excess starting material by preparative HPLC on a Waters 19x150mm C18 column eluted with methanol/water (45/65), and the solvent was removed on a rotary evaporator. The hydrochloride salt was recrystallized from 2-propanol/ether: 40 mg (62% based on KI); mp 227-230°C.
Radioiodination procedures
The effects of differing solvents, oxidizing agents and temperature upon the radioiodination yield were investigated. The general radioiodination procedure was as follows: to 6 µmol of precursor in a Reacti-Vial (Pierce Chemical Company) equipped with a magnetic stirrer there was added solvent, radioiodine in a minimum volume of aqueous base, and oxidant; aliquots of the reaction mixture at various times after addition of the oxidant were quenched with excess sodium metabisulfite (MBS); and the kinetics of the radioiodination were determined by radio-HPLC. These procedures helped to identify the best labelling routes which were then utilized for the larger scale radiosyntheses of DOI.
Radiosynthesis of 2,5-Dimethoxy-4-[125I or 123I]-Iodophenylisopropylamine
Method A. Via the N-trifluoroacetyl-(R)- or (S)-2,5-dimethoxyphenylisopropylamine.
To 300 µL of trifluoroacetic acid (TFA) there was added 1.8 mg (6 µmol) of (R)- or (S)-1-(2,5-dimethoxyphenyl)-2-trifluoroacetamidopropane ((R)-2 or (S)-2). Solutions of no-carrier-added 125I-iodide or 123I-iodide in 0.1N NaOH (5-50 µL) were added to the vial followed by 0.5 mg (2 µmoles) dichloramine-T (TCI Tokyo Kasei Organic Chemicals) in 20 µL TFA. The vial was sealed and the reaction mixture stirred at room temperature.
Aliquots of the reaction mixture were quenched with excess MBS at various times after the addition of dichloramine-T (DCT) to follow the progress of the reaction. The mixture was analyzed by reverse phase radio-HPLC utilizing a buffered methanol/water (60/40) eluent. The reaction mixture was quenched with 2 mg (10 µmoles) MBS when the radio-HPLC indicated greater than 90% incorporation of the radioiodine (approximately one hour). The solvent was evaporated at 50°C under a gentle stream of N2 and 300 µL of 2-propanol and 50 µL of 2N KOH were added. The pH of the solution was checked and additional KOH added if necessary to make the pH greater than 12. The vial was sealed and the mixture stirred at 50°C until deblocking was complete (>99% at ca. 30 min). Approximately 300 µL of water were added to the vial and the solution was filtered through a 0.45 µm Millipore filter prior to HPLC fraction collecting. A C18 analytical column was eluted with a buffered methanol/water solution (35/65), and the radioactive peak containing radioiodinated (R)- or (S)-DOI was collected. The solvent was removed by vigorously blowing N2 over the solution while heating in a 90°C water bath. When the product was contained in approximately 1 mL of solution, it was reinjected onto the C18 column with UV monitoring to determine its specific activity and was fraction collected as before. The solvent was completely evaporated and the product taken up in an ethanol/water (50/50) solution and stored at -20°C.
Method B. Via the N-phthalimide protected (±)-2,5-dimethoxyphenylisopropylamine.
To 300 µL of TFA there was added 1.9 mg (6 µmoles) of racemic (±)-1-(2,5-dimethoxyphenyl)-2-phthalimidopropane ((+)-5) prepared as previously described14. To this solution was added 5-50 µL of 125I- or 123I-iodide in 0.1N NaOH followed by 0.5 mg DCT in 20 µL TFA. The vial was sealed and the reaction mixture stirred at room temperature. The reaction was followed as before with radio-HPLC and quenched with MBS when the reaction yield exceeded 90% (1-2 h). The TFA was evaporated with a gentle stream of N2, and 300 µL butanol and 100 µL hydrazine hydrate were added. The vial was sealed and the mixture was heated to 110°C in an oil bath for approximately 5 min. An aliquot of the reaction mixture was analyzed by radio-HPLC to ensure that complete (>99%) deblocking had occurred. The mixture was taken up in a syringe and filtered through a 0.45 µm Millipore filter before chromatographing twice.
Method C. Via the unprotected (R)- or (S)-2,5-dimethoxyphenylisopropylamine.
To 300 µL of 2.0M phosphoric acid there was added 1.2 mg (6 µmoles) of (R)- or (S)-1-(2,5-dimethoxyphenyl)-2-aminopropane ((R)-1 or (S)-1) prepared as previously described3. To this solution was added 5-50 µL of 125I- or 123I-iodide in 0.1N NaOH followed by 0.4 mg (2 µmoles) of chloramine-T (CAT). The vial was sealed and the reaction mixture stirred at room temperature while following the progress of the reaction with radio-HPLC. The reaction was quenched with MBS when the reaction yield exceeded 90% (ca. 10 min). The mixture was made basic by the addition of NaOH and chromatographed twice with HPLC.
Results and Discussion
Table 1.
Comparison of the Effects of Precursors,
Solvents and Oxidantsa on the
125I-Radioiodination Yield of DOI.
Precursor |
Solvent |
Oxidant |
Yield at 2hb |
(R)-2 | TFA | DCT | 94 ± 6 |
(S)-2 | TFA | DCT | 91±5 |
(±)-2 | TFA | DCT | 94 ± 7 |
(±)-2 | TFA | DCT (0.5µmol) | 93 ± 5 |
(±)-2 | TFA | DCT (12µmol) | 5±2c |
(±)-2 | TFA | CAT | 91 ± 4 |
(±)-2 | TFAA | CAT | 83 ± 9 |
(±)-2 | HOAc | DCT | 23 ± 12 |
(±)-2 | HOAc | CAT | 3 ± 2 |
(±)-2 | HOAc/MeOH (25/75) | DCT | 2 ± 2 |
(±)-2 | MeOH | DCT | 9 ± 4 |
(±)-2 | HOAc | H2O2 | <1 |
(±)-2 | TFA | H2O2 | <1 |
(±)-5 | TFA | DCT | 91 ± 4 |
(+)-5 | TFA | CAT | 87 ± 5 |
(±)-5 | TFAA | CAT | 81 ± 4 |
(R)-1 | H2O/H3PO4 (2.0M) | CAT | 94 ± 3 |
(S)-1 | H2O/H3PO4 (2.0M) | CAT | 93 ± 5 |
(±)-1 | H2O/H3PO4 (2.0M | CAT | 93 ± 3 |
(±)-1 | H2O/H3PO4 (0.1M) | CAT | 66 ± 9 |
(+)-1 | H2O/H3PO4 (0.5M) | CAT | 78 ± 7 |
(±)-1 | H2O/pH 4.3 (0.5M) | CAT | 21 ± 5 |
(±)-1 | H2O/pH 7.2 (0.5M) | CAT | 3 ± 2 |
(±)-1 | TFA | DCT | 3 ± 2 |
(±)-1 | HOAc | DCT | 9 ± 3 |
(±)-1 | MeOH | DCT | <1 |
(±)-1 | HOAc | H2O2 | 14 ± 2 |
Notes:
- 6 µmol reactant, 300 µL solvent, and 2 µmol
oxidant unless otherwise stated. - Average of 3 or more separate reactions;
yield based upon the starting 125I-iodide. - 12 µmol DCT yielded 65% at 10 min, but
the product was converted into another
unidentified compound at later times.
Summaries of the radiochemical yields and specific activities of the 125I- and 123I-labelled DOI products are presented in Tables 2 and 3. Use of 1 resulted in the highest isolated yields using a 2M phosphoric acid solution. The N-phthalamide derivative gave crude radioincorporation results similar to 2, but recovery of the hydrolysis product was not as efficient. There were no significant differences in the specific activities of the DOI products produced by the three radiosynthesis methods.
Abbreviations Used in Text
- CAT
- Chloramine-T
- DCT
- Dichloramine-T
- DOB
- 4-Bromo-2,5-dimethoxy-
phenylisopropylamine - DOC
- 4-Chloro-2,5-dimethoxy-
phenylisopropylamine - DOI
- 2,5-Dimethoxy-4-iodo-
phenylisopropylamine - DOM
- 2,5-Dimethoxy-4-methyl-
phenylisopropylamine - MBS
- Sodium metabisulfite
- TFA
- Trifluoroacetic acid
- TFAA
- Trifluoroacetic anhydride
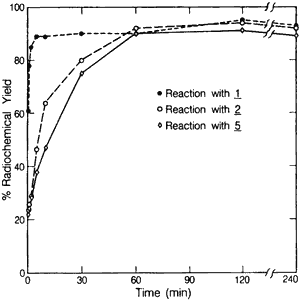
Figure 1.
Crude 125I-radiolabelling yields; Method A, B, and C.
The kinetics of three methods are compared in Fig. 1. Radioiodination of 1 resulted in yields of 90% in 5 min, while 2 and 5 reached maximum values of approximately 90% at 1 h. Further reaction of solutions containing 2 and 5 resulted in a 10% loss of product over 48 h and the formation of another unidentified radiolabelled compound which eluted after 4 with the reverse phase HPLC conditions utilized here. The rates of the reactions were not greatly affected by a temperature increase to 70°C (data not shown).
All three labelling methods resulted in the crude radioincorporation yields of approximately 90%. Radioiodination of the free amine (1) provided faster labelling kinetics and greater overall yields due to the simpler synthesis which did not involve deprotection. Total radiosynthesis time with this method was 2-3 h.
Table 2.
125I-Iodination: Yields and Specific Activities
{kind=link}
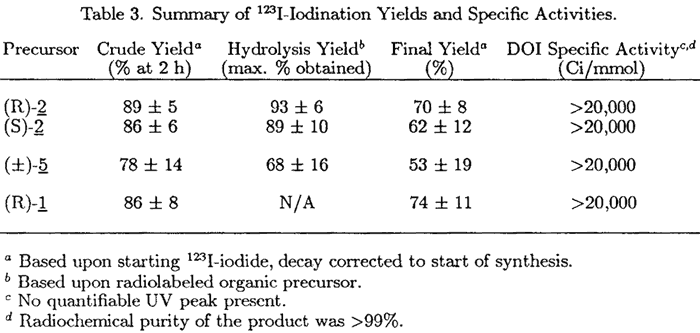
Table 3.
123I-Iodination: Yields and Specific Activities
{kind=link}