05-31-03 14:15
No 436852
Hello!
I've been interested in psychoactive substances for some years but never been able to solve this task.
Cheap converting amides(-CO-NH-) to amines (-CH2-NH-). I've tried LiAlH4 in ether or hydrocarbon medium but it seems to be very expensive.
Do someone know how to do this cheap?
And another one, the 3D formulas where the N in sp2 is connected to sp2 carbon in TIKHAL are not exact.
The sp2 Nitrogen forms 3 valen bonds with the sp2 electrons and with the 2p z. where the electron couple is situated makes p-pi interaction with the pi bond of the sp2 carbon, so the structure is planar (like NH2-NO2) and not triangle piramid. Anyway, it can turn to triangle pyramid if you heat the compound.
For more info on the subject see urea quantum structure.
Don't drink and drive, but smoke and fly!
(Moderator)
05-31-03 16:57
No 436869
The usual method besides using LAH in ether is catlytic hydrogenation using copper chromite catalysts, at 200 to 300 atmospheres and 250 to 265 °C, yields are often low due to side reactions, cleavage et cetera
Chemistry is our Covalent Bond
(Stranger)
05-31-03 18:47
No 436887
A must have article for electrolytic reduction:
'Electrolytic Reduction of Organic Compounds' Chemical Reviews vol 62 pages 19-40 (1962)
Electrolytic reduction of amides to amines:
Zhurnal obshchii khimii (J. Gen. Chem. USSR) 17, 1651 (1947)
Zhurnal obshchii khimii (J. Gen. Chem. USSR) 11, 51 (1941)
Bull. Chem. Soc. Japan 11, 41 (1936)
Trans. Electrochem. Soc. 84, 165 (1943)
Catalytic hydrogenation of amides to amines:
JACS 56, 247 (1934)
JACS 61, 3449-3502 (1939)
Patent US2187745
(Hive Addict)
06-01-03 03:35
No 436954
Many amides can be reduced to amines by triacetoxyborohydride formed in situ from sodium borohydride and glacial acetic acid.
This method is sufficient for many amides:
To a suspension of the amide, toluene and sodium borohydride is added GAA dropwise without cooling. When the acid has been added the mixture is allowed to stir for one hour with or without heating. The reaction progress is checked with TLC. Destruction of excess hydride and standard A/B workup gives amines in the range of 40-80% of the theoretical yield. In many cases trifluoroacetic acid gives better yields than acetic acid.
Freaky
(Stranger)
06-03-03 18:11
No 437675
All amides are not alike. Some are harder to reduce than others. Some are very resistant to reduction. N,N-dialkylamides are easier to reduce electrolytically than N-alkylamides and N-alkylamides are easier to reduce than non substituted amides. Get that Chem. Rev. article and read it.
(Moderator)
06-07-03 18:42
No 438543
(Rated as: excellent)
'Electrolytic Reduction of Organic Compounds' - Chemical Reviews, Vol 62, pp 19-40 (1962) (../rhodium/pdf /electroreduc
A. Introduction to Electroorganic Reduction
1. Cathode reactions
The passage of electricity through two electrodes immersed in an electrolyte results in the deposition of a metal or the evolution of hydrogen from the cathode. Reducing conditions exist at the cathode of sufficient power to enable the electrolytic hydrogen to add to unsaturated organic molecules or to remove oxygen from organic molecules. Unlike the reduction of most inorganic substances, the reduction of organic compounds is sometimes reversible (e.g., quinone-hydroquinone, azobenzene-hydrazobenzene, nitrosobenzenephenylhydroxylamine). Definitions of many of the terms used are included in earlier reports on electrochemistry (323) and are not discussed here.
2. Theories of reduction at the cathode
Several theories have attempted to account for the reducing action of the cathode. The most reasonable theory, first proposed by Haber (130) and later supported by Creighton (55), states that hydrogen atoms are released at the cathode by the discharge of a proton with an electron. The atomic hydrogen thus formed reacts with the organic compound.
The hydride ion has been considered to be the active reducing agent (109). Other workers (51, 368) believe electrolytic reduction to be a phenomenon of simultaneous electrolytic and chemical reduction. Certainly, the electrolytic reduction of an organic compound in a basic solution at a mercury cathode could conceivably be due to the chemical activity of the electrolytically formed amalgam. There may also be the phenomenon of simultaneous electrolytic and catalytic reduction; the activity of platinized platinum and nickelized nickel cathodes probably is due to the catalytic effect of the finely divided cathodic surface. Evidence has been presented (171, 198) that reduction may take place by direct transfer of electrons from the electrode to the substance being reduced; this hypothesis presumes the addition of the highly mobile proton to a negative center. It has been observed (342) that an unstable, benzene-soluble, reducing substance is formed at platinum or lead cathodes. This unknown substance was shown not to be a gas, tritium, or ferrous ion; it was not, however, further identified. Some investigators (115, 186, 337) have stated that the cathode may enter into the electroorganic reduction; they have reported the formation of organometallic compounds, particularly when carbonyl groups were reduced at lead or mercury cathodes.
It appears that to a large extent the mechanism of electroreduction depends upon many factors. In the case of the electroreduction of diphosphopyridine nucleotide (167), it appears that the process involves proton transfers, rather than a direct transfer of hydrogen atoms, since no dependence is found on the use, of a variety of electrodes of different overvoltages. Antropov (16) has presented a mathematical treatment which supports the theory that nonpolar groups of compounds such as phenol, acetylene, vinylacetylene, crotonic acid, oleic acid, and sorbic acid are reduced on low-overvoltage cathodes by hydrogen atoms adsorbed on the electrode surface, while polar groups of acetone, acetaldehyde, 1-acetyl 2-propanol, acetophenone, benzophenone, salicylaldehyde, and oxalic acid are reduced on cathodes of high overvoltage by hydrogen ions activated in the double layer. Many other papers have appeared which discuss the mechanism of electroorganic reduction in specific systems (140, 218).
3. Disadvantages and advantages of electroorganic reduction
The two main disadvantages which have been offered for electroorganic reductions (116, 320) are that the reductions are usually of a low velocity and that the process requires careful control of many conditions.
There are several apparent advantages of electroorganic reduction:
(1) impurities from a reducing agent are not present in the reaction mixture;
(2) partial reduction may be achieved by altering the reduction conditions;
(3) selective reduction of certain unsaturated groups in the presence of other groups has been acomplished (208);
(4) electroorganic reductions have been successful in the presence of compounds that usually poison catalytic reductions, and such substances as mercury, halides, hydrogen sulfide, and arsenic sulfide have been demonstrated to be electrocatalytic (163). Electroorganic reduction can also be developed into procedures suitable for large-scale preparative work (80).
[u]B. Factors That Influence Electroorganic Reduction
1. The cathode potential
It has been stated (208) that the cathode potential is the factor that determines the success or failure of electroorganic reductions. This fact has long been recognized and conceded by responsible investigators, but very few have described the cathode potential used during reduction. Rather, a host of less important factors have been enumerated and described.
Haber (129, 130) first recognized the importance of controlled cathode potential for electroorganic reductions. He showed that in an alkaline solution at a platinum cathode nitrobenzene was reduced in good yield to pure azoxybenzene when the cathode potential was held at 0.9 v.; a potential of -1.3 v. gave good yields of pure hydrazobenzene. This use of cathode potential has been found to lead to purer products in many cases. In the electroreduction of 8-chlorotheophylline (353), to theophylline the cathode potential was varied until the potential which gave the purest product was found. At all other potentials mixtures of products were obtained. Acetylene has been selectively reduced (32) to ethylene or ethane at a platinum cathode by controlling the cathode potential. Other workers also have stated (10, 12, 80, 207) that the cathode potential was of primary importance for controlled electroorganic reductions, and additional examples will be found laterin this review. The electroreduction of nitrobenzene at an empirically established cathode potential of about -1.5 v. has been studied at a number of different cathodes (210). In all cases about 60 per cent yields of azoxybenzene and 40 per cent yields of aniline were obtained at zinc, copper, platinum, tin, and nickel cathodes.
The above work has been reiterated and pushed even further by the determination, polarographically, of the optimum cathode potential for the reduction of 9-(o-iodophenyl)acridine to 9-(o-iodophenyl)dihydroacridine at a mercury cathode (208). Although all previous methods and attempts to carry out this reduction had failed, because of an excessive reduction that removed the iodine atom, the use of the polarographically determined, constant cathode potential gave the desired product quantitatively. Although other workers (165, 256) also have shown that the potential found in polarographic work is confirmed by electroreductions, it has been pointed out (80, 92) that polarographic data alone do not rigidly demonstrate the efficacy of a preparative method; therefore polarographic data have not been included in this review (see reference 360 for a review of such data).
2. The cathode overvoltage
Two definitions of overvoltage have been presented (58):
(1) the overvoltage of a gas upon a specific electrode is the polarization involved in the evolution of that gas on that cathode at a specified current density;
(2) the overvoltage of a gas on a specified electrode is the minimum polarization at which visible evolution of gas occurs, or at which there is a marked increase in current density.
The higher the overvoltage value of any cathode, the greater is the reducing power of that cathode toward any organic depolarizer. Tafel (333, 335) was the first to recognize the relation between high overvoltage and a high reducing power. One of the early examples of this relationship was the inability of a smooth platinum cathode of low overvoltage to reduce cinnamic acid to hydrocinnamic acid; this reduction was accomplished, however, at the high-overvoltage lead and mercury cathodes.
The concept of overvoltage is important, since in an electroorganic reduction when a depolarizer is present, the potential at which hydrogen is evolved as a gas from the cathode marks that potential as the critical point above which the hydrogen is not absorbed by the depolarizer. Below this potential the atomic hydrogen reacts with the depolarizer as fast as it is formed.
Various theories of overvoltage have been discussed in great detail (118). The most acceptable view is that the metals of lower overvoltage function as catalysts for the reaction H+ + e- H0 by decreasing the energy of activation for the process. Metals of high overvoltage have a large energy of activation, and hydrogen atoms formed at a metal of high overvoltage have a much greater potential energy than those liberated at metals of low hydrogen overvoltage.
Considerable study of overvoltages of various metals has demonstrated that the absolute values obtained have little import; only the relative differences in hydrogen overvoltages are of significance. Many of the existing data on overvoltage have been summarized (58, 183). The average overvoltage, obtained by averaging various literature values (49, 58, 183, 335), of hydrogen on various cathodes. It should be reemphasized that the absolute values given are of no significance; only the approximate order of arrangement of the cathodes should be considered.
The following factors have been listed as those which affect the overvoltage of hydrogen on metals (58):
(1) an increase in current density decreased the overvoltage;
(2) an increase in temperature decreased the overvoltage;
(3) an increase in pressure decreased the overvoltage;
(4) the overvoltage increased with time;
(5) an alternating current superimposed on a direct current lowered the overvoltage;
(6) the overvoltage (116) is less in an alkaline solution than in an acid solution.
Chemistry is our Covalent Bond
(Moderator)
06-07-03 18:48
No 438544
3. Cathode material and condition
The cathode material and its condition are rather intimately connected with the cathode potential and cathode overvoltage, but several observations on cathode materials properly belong by themselves. In addition to the more specific property of cathode overvoltage, the cathode material and condition have an effect upon electroorganic reductions.
The purity of the cathode is of prime importance. It has been demonstrated (333, 334) that when lead is used as a cathode, its reducing power is nil when there is present as little as 3 mg. of copper, 0.5 mg. of silver, or 0.04 mg. of platinum per square decimeter of lead surface. The difference between 99.9 per cent lead and 99.99 per cent lead is often enough to cause failure in an electroorganic reduction (320). All leads, tabs, and connections of a metal different from the cathode were shown to be deleterious to the cathode activity.
The physical state of the cathode may often play a leading role in an electroreduction. Generally a rough cathode has a lower overvoltage than a smooth, one; but since it also has a greater surface area, the depolarizer has more opportunity to react with atomic hydrogen on it. Cinnamic acid failed to undergo reduction at a smooth platinum cathode but reacted at a platinized platinum cathode. It was found (203) that the rate of cathodic reduction of compounds such as aconitic acid and quinoline was increased by continuous renewal of the mercury surface, while the rate of reduction was decreased with naphthalene and anthracene. In these experiments, the surface of the mercury was renewed by motion, and at a high rate of motion reduction was stopped in many cases. It was concluded that if the mercury had a catalytic effect, renewal accelerated the reduction; if the reduction were due to
electrolytically produced amalgam, then renewal counteracted the reduction; and finally that if renewal had no effect, as is the case of sorbic acid, then the reduction was truly electrolytic and depended only on the cathode potential.
The history of a cathode is of importance. A case has been cited (320) in which commercial cadmium, presumably of a microcrystalline character, was unfit for use as a cathode until melted and recast. Cathodes which have been used previously as anodes have been shown to have special activity as cathodes.
In addition to the above observations, it has been ascertained that certain cathodes appear to have a definite catalytic effect upon certain reactions which is unexplained in terms of cathode potential or overvoltage. Only platinized platinum, the cathode of lowest overvoltage, reduced aromatic nuclei (24, 25, 102). At a copper cathode, unlike other cathodes, nitrobenzene was reduced completely to aniline in both acid and alkaline solutions.
4. Current density
Current density is usually defined as the amperes of current applied per square decimeter of cathode surface. Too much importance has been placed upon the influence of current density in electroorganic reductions. Its importance lies primarily in the fact that it is a means of controlling the cathode potential. Usually the current density is the major experimental point in the literature (it is sometimes even listed in papers which do not mention what the cathode material is), but it has been shown (10) that the current density is of little value in reproducing work unless everything else is reproduced exactly, and that quite often electroorganic reductions are somewhat independent of current density and applied voltage.
5. Solvent, pH, and concentration
Because of the water insolubility of most organic compounds, a solvent must be used that conducts an electric current and also dissolves organic compounds. Generally this end is attained by adding acetone, alcohol, or lower-molecular-weight organic acids to a water solution of the electrolyte. Organic solvents raise the resistance of an electrolyte and lower the overvoltage of the cathode (116). Another disadvantage is the possibility of reaction of the organic solvent either at the cathode, with the depolarizer, or with the reduction product.
Use of concentrated water solutions of sodium and potassium aromatic sulfonates to increase the solubility of the depolarizer has been found to be of advantage (214, 215). Suspensions of the depolarizer in the catholyte have in many instances been employed successfully. Dispersion has been aided by use of a cellulose ether (278). It has been demonstrated (260) that surface-active agents, as emulsifying aids, decrease current efficiencies during electroorganic reductions.
When the reducible group forms the negative part of a dipole the lowering of the dielectric constant of the solvent, as in the addition of dioxane to water (108), forces the reducible group away from the cathode and thus lowers the reduction efficiency. This has been found to be true with p-nitrophenol and nitrobenzene; however, with benzaldehyde and phthalimide the increase in solubility overbalances the change of dielectric constant and the reduction efficiency is increased (108). Other workers (358) have pointed out the importance of the polarity of the organic compound being reduced and emphasized that only the dipole moment of the group being reduced is important. A compound can be reduced if its dipole moment is greater than that of the solvent.
The hydrogen ion concentration of the catholyte plays an important role in electroorganic reductions. The course of a reduction may be controlled by altering the pH, and the formation of intermediate condensation products may be accelerated with certain pH conditions. As examples of the first effect:
(1) it has been shown (268) that the reduction of carbon dioxide to formic acid takes place only in a very strong acid;
(2) aromatic carboxylic acids are reduced to benzyl alcohols in acid solution (224), whereas in alkaline medium a nuclear reduction occurs; and
(3) reduction of levulinic acid gave the hydroxy acid in acid solution and valeric acid in a basic solution (338).
The reduction of nitrobenzene in a basic solution leads to products with two aromatic rings (89, 90, 137, 211), presumably formed by a base-catalyzed condensation of such intermediates as nitrosobenzene and phenylhydroxylamine. The formation of bimolecular products from the reduction of carbonyl compounds in acid solutions may also be cited (91, 116, 117, 164, 184, 185) as examples of the effect of the pH of the reduction solution.
Generally it has been observed (116, 118) that the rate of reduction is higher in a high concentration of depolarizer than in a low one. High concentrations of depolarizer often give a more rapid reduction, but no increase in yields (84). The effects of the type of compound being reduced can be seen in Section III of this review; compounds which contain unsaturated nitrogen are generally the easiest to reduce, while aromatic benzenoid compounds are by far the most difficult to reduce.
6. Temperature
Temperature influences three factors (116): the cathode overvoltage, the rate of reduction, and the rate of diffusion of the depolarizer to the cathode. In addition, certain side reactions can be influenced by the temperature. The cathode overvoltage loss in most
metals amounts to about 0.02-0.03 v. for every tendegree rise in temperature. Despite this drawback, the rate of reduction in some instances is so low at room temperature that heat is necessary to bring about reduction (267, 336, 339, 359). The increase in reduction, or velocity of reduction, with heat probably is caused by an increase in the rate of diffusion of the depolarizer to the cathode (184). The heating of the cathode itself has been found to be of no value in at least one case (228).
7. Catalysts
Catalysts for electroreduction can be divided into three types (116, 119):
(1) salts of metals such as copper, tin, lead, and mercury which plate out on the cathode, thereby altering its nature and catalytic action;
(2) salts of metals such as titanium and vanadium, which are reduced at the cathode from a higher to a lower valence state and in this latter state reduce the organic material;
(3) finely divided metals with a true catalytic surface, either suspended in the catholyte or fixed to the cathode surface. The influence of catalyst poisons has been studied (295), and it has. been found that the addition of arsenic trioxide increased the yields of aniline, but decreased the yields of glyoxalic acid from oxalic acid in electroreduction. From this it was deduced that the reduction of nitrobenzene was based on atomic hydrogen, but that the latter reduction depended on the hydrogen overvoltage.
8. Agitation and miscellaneous factors
As has been mentioned, the rate of diffusion of a depolarizer to the cathode during reduction limits the velocity of reduction. If electrons are supplied to protons faster than the atomic hydrogen is used to saturate the depolarizer, a state of concentration polarization may be attained and gaseous hydrogen evolved. It has been shown (184, 229) that the reaction velocity depends upon the diffusion of the depolarizer to the cathode. In a stirred solution, however, one can reach a point where the velocity of reduction is independent of the rate of diffusion (184).
In a few cases (242, 251) electroorganic reductions have been carried out under a pressure of either hydrogen or carbon dioxide. In many cases the yields are increased at increased pressure, but the results have been variable.
The use of ultrasonics (181) in the reduction of nitrobenzene has been found to give a ten to fifteenfold increase in the limiting current.
Chemistry is our Covalent Bond
(Moderator)
06-07-03 18:50
No 438545
C. METHODS AND APPARATUS FOR ELECTROORGANIC REDUCTIONS
1. Use of diaphragms
The organic depolarizer, cathode, and catholyte must be isolated from the anode section of the electrolytic cell; otherwise the organic compound could diffuse into the anolyte and be oxidized. In most cases this end is accomplished by enclosing or separating the cathode section of the electrolytic cell from the anode section with a partition which conducts an electric current but prevents diffusion of the depolarizer to the anode. Partitions have been constructed of porous clay, alundum, wood, sintered glass, and paper. Because of the high resistance of partitions and their tendency to become impervious, as a result of the formation of tar, such diaphragms are but a poor solution to the problem of anodic oxidation. Many attempts to solve this problem by rendering the anode passive to the depolarizer have met with failure. However, several investigations (95, 96, 265, 277, 298, 299) have been reported wherein an effective separation of the catholyte was accomplished by using either two immiscible solutions, or solutions of considerably different specific gravity. In these cases, of course, the cathode and anode are arranged one above the other. It has been reported (230) that no diaphragm was needed for the reduction of fumaric acid, whereas reduction of maleic acid required a diaphragm.
2. Cathode materials
Cathodes may be prepared from metallic sheets, wire, gauze, rods, or wool. The materials from which cathodes have been made are platinized platinum, platinum, tungsten, antimony, gold, nickelized nickel, nickel, tantalum (386), palladium, silver, carbon, tin, copper, iron, graphite, bismuth, aluminum, mercury, zinc, lead, lead dioxide, gallium (371), cadmium, thallium (385), and amalgams of several of these metals. Platinum, mercury, and lead appear to be the most widely used cathodes. In many instances the pH of the catholyte governs the choice of the cathode. Although, as mentioned earlier, the purity of the cathode has been shown to be of prime importance, there have been several instances reported in which alloys, amalgams, and plated cathodes have proven of value. The use of the following cathodes has been reported: iron-nickel alloys (115), cadmium-bismuth alloys (263), Monel (215), bronze (215), and amalgamated cadmium. Cathodes have been studied in which-, tin was plated on copper and lead; lead, copper, zinc, nickel, iron, and silver were plated onto porous graphite; and nickel, tin, and copper were plated onto nickel.
S. The catholyte
The catholyte may consist of either an aqueous or a nonaqueous medium. In the event water is the catholyte, insoluble organic compounds are brought into solution by adding such solubilizers as methanol, ethanol, formic acid, or acetic acid. Concentrated aqueous solutions of sodium or potassium benzene-, toluene-, or xylenesulfonates have been shown (215) to be excellent hydrotropic agents. Some instances have been reported in which the insoluble organic compounds were emulsified in water by adding Igepon T, Nekol BX, sodium lauryl sulfate, cetylpyridinium bromide (260), or a cellulose ether (278).
In order to make aqueous solutions more conductive to an electric current, acids such as sulfuric acid, hydrochloric acid, hydrocyanic acid, and ammonium hydrogen sulfate have been utilized. A basic conducting medium has been attained by adding ammonium hydroxide, sodium hydroxide, or sodium carbonate to water. Nearly neutral aqueous solutions have been prepared by adding ammonium chloride, ammonium sulfate, or sodium acetate to water. Methanol, ethanol, formaldehyde, formic acid, acetic acid, ethyl acetate, hydrogen cyanide, liquid ammonia, and fuming sulfuric acid have served as nonaqueous catholytes. Sodium and potassium acetates have been added to many of the above catholytes to increase the conductivity of the electrolyte.
4. The anode and the anolyte
The anode should be a nonattackable electrode such as platinum, carbon, lead, nickel, or iron. Generally, the anolyte should be of the same material and concentration as the catholyte, for this reduces to a minimum complications that may arise because of diffusion through the diaphragm.
5. Source, measurement, and control of current and potential
Six-volt storage cells are the best source of power for electroorganic reduction. The consumption of current may be measured by an ammeter, and the potential drop across the cell by a voltmeter. A variable resistance must be included in the circuit in order to control the current density at the cathode. Many varieties of power packs and converters are also now available.
The determination of the quantity of electricity (coulombs) passed through a cell may be accomplished by recording accurately the time intervals and the amperes. An easier method is the utilization of silver, copper, or gas coulometers. The silver or copper coulometers are less desirable for routine work, for the gas coulometer visibly evolves gas in direct proportion to the current passed through the reduction cell, and may be referred to with ease as the reduction proceeds. In some investigations the cathode of the reduction cell itself is enclosed so that any gas escaping from it may be measured. If the gas evolved by the coulometer and by the cathode is known, the absorption of hydrogen by the depolarizes may be determined easily. Almost all references contain some description of the apparatus and many include wiring diagrams. The above apparatus has been described in- an excellent manner (260).
A determination of cathode potential requires the use of a probing electrode, potentiometer, and standard cell; a description of this apparatus has been presented (208). Designs of several pieces of apparatus which can be used to maintain constant cathode potential have been described (3, 107, 207). An apparatus to maintain constant cathode potentials as high as -6 v. has been described in both a micro and a macro form (10). A simple potentiostat, which requires no preliminary adjustment or calibration and which is sensitive to ±0.01 v. up to 25 v., has been reported (207).
6. Control of temperature
The temperature of a cell may be controlled by placing the entire cell in an ice bath, on a steam cone, or in a constant-temperature bath. A glass coil in the catholyte may be used to circulate cold or hot water; occasionally the cathodes themselves are hollow tubes through which cold or hot water circulates.
7. Agitation
Stirring may be accomplished by the usual types of paddle stirrers, by rotating a cylindrical cathode, or by circulating the liquid by means of a small pump. In some cases bubbling of air or other gas through the catholyte provides sufficient agitation.
Reduction of carbon-carbon unsaturated compounds
Ethylenic bonds
Carbon-carbon unsaturation, which is found in isolated ethylenic bonds, is very resistant to electroorganic reduction. The only successful attempts to reduce isolated ethylenic bonds have utilized cathodes with a spongy surface (for example see reference 258). It is to be questioned whether these reductions are bona fide electrolytic reductions, or merely reductions by molecular hydrogen at a catalytic surface.
Carbon-carbon double bonds conjugated with either another ethylenic group, a carbonyl group, or a nitro group appear to be easier to reduce than isolated ethylenic bonds. Either the use of high-overvoltage cathodes, such as lead or mercury, or of an activated cathode surface, such as platinized platinum or nickelized nickel, is necessary to bring about this type of electrolytic reduction. The diene acid B,y-diphenylmuconic acid, however, is reduced only to B,y- The olefinic group in acids with a double bond between a benzene ring and the carboxyl group, or between two carboxyl groups, can be reduced using lead or mercury cathodes. In acidic solutions at a mercury cathode certain acids undergo bimolecular reduction to adipic acids (172), but this is not general (374). The ,electrolytic reduction of 3-nitrocinnamic acid to B3-nitrophenylpropionic acid (293) was very unusual, for the nitro group is generally more easily reduced at the cathode than is the ethylenic group. Reduction of dimethylmaleic and dimethylfumaric acids at a mercury cathode gave racemic acid and meso-a, a'-dimethyl.succinic acid, respectively. This is apparently the first clear case of trans addition at an electrode (266).
Chemistry is our Covalent Bond
(Moderator)
06-07-03 18:53
No 438546
2. Aromatic unsaturation
A number of carbon-to-carbon double bonds in nitrogen heterocyclic compounds, including some alkaloids, have been reduced. Generally, but not always, either high-overvoltage cathodes or especially activated cathodes were necessary to bring about these electroreductions. Aromatic rings containing a nitrogen atom were reduced with relative ease at high-oversoltage cathodes, particularly lead. A comparison of the activity of heterocyclic and homocyclic rings may be noted in the reduction of quinoline to 1,4-dihydroquinoline and 1,2,3,4-tetrahydroquinoline (200).
Platinized platinum cathodes were required for the complete reduction of the benzene nucleus. Claims that aromatic carboxylic acids were completely reduced in the nucleus under basic conditions at lead or mercury cathodes (223) appear to be ill-founded (224). Several 1-alkoxynaphthalenes have been reduced to 3,4dihydronaphthalenes by use of a tetrabutylammonium iodide catholyte at a mercury cathode maintained at a constant potential (80).
3. Acetylenes
Nickelized nickel, platinized platinum, and silverplated copper cathodes served adequately for the reduction of acetylenes to ethylenes or saturated hydrocarbons. Acetylene can be reduced to ethylene by controlling the cathode potential (32). In a number of cases ethynes have been reduced to cis olefins (47).
B. REDUCTION OF CARBON-NITROGEN UNSATURATED COMPOUNDS
Carbon-nitrogen unsaturation has been reduced under electrolytic conditions in a variety of compounds. Ketoximes and aldoximes have been reduced to amines in acid solution, generally at a lead cathode.
The reduction of imino ethers to amines involves not only the saturation of a carbon-to-nitrogen double
bond, but also the hydrogenolysis of an ethoxy group to give ethanol (362).
Reduction of Schiff bases to secondary amines and related compounds has been carried out at high-overvoltage cathodes of lead or mercury in an acid solution. With some Schiff bases derived from ketones the use of a lead cathode and an alkaline solution has been reported (149). The reduction of a nitro compound in the presence of a carbonyl-containing compound may also lead to a secondary amine, the Schiff base probably being an intermediate.
Carbon-nitrogen unsaturation in heterocyclic compounds has also been reduced electrolytically, in many cases at the same time as carbon-carbon unsaturation.
A lead cathode is generally necessary for the reduction of the carbon-to-nitrogen triple bond. Unlike the other carbon-nitrogen compounds, a neutral solution appeared to be the most favorable for the reduction of cyanides to amines. It has been shown (235) that yields of amines by electroreduction of cyanides fell off as the catholyte was made either acidic or basic. In some cases, however, an acidic solution has been used successfully (239). A cyano group attached directly to a benzene ring gives good yields of a benzylamine upon electroreduction, but benzyl cyanide is hardly reduced at all to the corresponding amine (235) (however, see reference 166).
C. REDUCTION OF CARBONYL COMPOUNDS
1. Aldehydes
The electrochemical reduction of carbonyl compounds has been studied almost as extensively as has that of the nitro group. The reduction of saturated aliphatic aldehydes may take several routes. Monomolecular reduction appeared to be favored by an acidic catholyte. Use of a mercury or lead cathode generally gave an alcohol, while at a cadmium cathode, of highest overvoltage, reduction of an aldehyde to a hydrocarbon may take place. An example of bimolecular reduction is the reduction of glyoxylic acid to tartaric acid at a lead or mercury cathode in alkaline, neutral, or acid solution (42). Conjugated unsaturated aldehydes were more easily reduced than the isolated carbonyl groups.
As with the aliphatic aldehydes, the electrolytic reduction of aromatic aldehydes may take several courses: conditions for the electroreduction of aromatic saturated aldehydes. An acidic catholyte and high-overvoltage cathodes appeared to be most desirable, although good yields also were reported from 'basic solutions. It has been stated (287) that copper, the' cathode of lower overvoltage, favors bimolecular reduction.
2. Ketones
The electrolytic reduction of keto groups to methylene groups in aliphatic compounds has been reviewed by Swann (324). This reduction generally takes place only in acid solution; an amalgamated zinc cathode appears to be the most active. Reduction of aliphatic ketones at cathodes of high overvoltage, generally in alkaline solution, tends to lead to pinacol formation, although this reaction is not general for all aliphatic ketones. A host of
other products have been isolated from the electroreduction of aliphatic ketones. Generally the reduction of aldehydes can be accomplished a little more readily than the reduction of ketones.
Acetone has been reduced to isopropyl alcohol and pinacol in aqueous solution at a mercury cathode (141). It is believed that the acetone is first adsorbed on the mercury, forming a covalent bond. This may then dissociate as a free radical to form pinacol, or may first add a proton and then dissociate from the mercury surface as an ion, which adds a second proton to give the alcohol.
In certain cases an electroreduction of an ethylenic bond in an aliphatic unsaturated ketone may occur selectively. Also, with a, l3-unsaturated ketones, a bimolecular reduction may occur (187).
Reduction of saturated aromatic ketones at a lead cathode in slightly basic solutions gave high yields of secondary alcohols. An acidic catholyte generally produced a mixture of the pinacol and the benzhydrol; if the catholyte was strongly acidic, rearrangement to the pinacolone sometimes occurred. o-Nitrobenzophenone was reduced in alkaline solution at a lead cathode to o-hydroxylaminobenzohydrol, which loses water to form 1,2-dihydroanthranil (23.) To reduce aromatic ketones to hydrocarbons it is generally necessary to use amalgamated zinc cathodes. The reductive coupling of 4-methoxyacetophenone with 4-amino- and 4dimethylaminoacetophenones to yield mixed pinacols is achieved by electrolysis at a mercury cathode (14). It is of interest to note that 2,3-bis(p-aminophenyl)2,3-butanediol can be obtained only by electrolytic reduction of the appropriate ketone in an aqueous hydrochloric acid medium at a mercury cathode (10, 11). The usual chemical methods gave only the carbinol. It is also of interest to note in regard to the reduction of p-aminoacetophenone that using an aqueous hydrochloric acid solution at a mercury cathode with a reference potential of -1.1 v. gave the carbinol, while a reference potential of -1.5 v. (10, 11) or a tin cathode (196) gave the pinacol. A number of Mannich bases also have been reduced to pinacols (12). Depending upon the reference potential used, either a low-melting or a high-melting form of the pinacol was obtained from p-dimethylaminoacetophenone (5).
An acid solution with a high-overvoltage cathode, such as lead or mercury, is best for the reduction of cyclic ketones. Alcohols, pinacols, pinacolones, and hydrocarbons have been isolated from the electroreduction of cyclic ketones. Reduction of 2-methylcyclohexanone at a mercury or lead cathode gave pure trans-2-methylcyclohexanol, at a nickel cathode a mixture was obtained, at a copper cathode pure cis-2methylcyclohexanol was obtained, and at a platinum cathode no reduction took place (18). The reduction of 2-ethyl-l-methyl-3-piperidone has been reported to give products such as 1-methyl-2-propylpyrroline, 2-ethyl-3-hydroxy-l-methylpiperidine, N-methylheptylamine, and 1-methyl-2-propyl-2-pyrroline (193) depending on the cathode and the temperature. Thus reductions, and rearrangements of the Clemmensen type, can be obtained by electrolysis. Reduction of keto acids and keto esters may take several courses. Generally an acidic catholyte allowed complete reduction to hydrocarbons with hydrogenolysis of the ester group. A basic catholyte usually gives a less reduced product than an acidic catholyte.
Quinones are reduced easily at the cathodes of lowest overvoltage in acid solution. For example, the following gave the corresponding hydroquinones: anthraquinone (244), 3,6-bis(1,4-dihydroxyphenyl)-1,4benzoqu
3. Carboxylic acids, esters, and lactones
An acid solution at a lead or mercury cathode was preferred for the electroreduction of aliphatic carboxylic acids to aldehydes, alcohols, or hydrocarbons. With these cathodes, ethyl oxalate in ethanolic sulfuric acid has been reduced to ethyl glyoxylate ethyl hemiacetal (252). T
Aromatic carboxylic acids are reduced to alcohols in a sulfuric acid catholyte using a lead or mercury cathode. It is possible to reduce certain aromatic acids to aldehydes in alkaline solution in the presence of the borate ion at a mercury cathode (227). Isonicotinic acid has been reduced to 4-picoline at an amalgamated lead cathode (364). Aromatic carboxylic acid esters have been reduced to the corresponding ethers or alcohols in an acid catholyte at mercury or lead cathodes.
Chemistry is our Covalent Bond
(Moderator)
06-07-03 18:54
No 438548
4. Amides, imides, and related compounds
The reduction of amides to amines has been carried out in acidic catholytes (some aliphatic amides have been reduced in basic solutions) at lead cathodes. The more substituted the amide nitrogen, the better the yield of amine; a phenyl group attached to the amide group increases the yield of substituted benzylamine. In fact, any aryl group linked directly to the carbonyl function gave good results, indicating that the greater the electron-donating effect of substituents on the amide nitrogen and the a-carbon atom, the greater the ease of reduction. A few instances of hydrogenolysis of the nitrogen compound to give ammonia or a simple amine have been reported. Attempts to reduce p-aminobenzanilide have been without success at many cathodes (38).
Imides of dibasic acids have been reduced to cyclic lactams and cyclic amines in acid solution. Generally a lead cathode is used, but cadmium and amalgamated zinc have been useful in some cases. Occasionally the ring has been opened by hydrogenolysis. In the case of many aliphatic imides the product from the reduction of only one carbonyl group may be isolated in good yields, while electroreduction of both carbonyl groups gave poor yields of the corresponding cyclic amine. N-substituted tetrachlorophthalimide can be reduced to a hydroxyphthalimide at a palladium cathode and a potential of -0.68 v. This hydroxyphthalimide can then be reduced further to an isoindoline at a reference potential of -1.10 v. (13). The mechanism of this reduction has been discussed in some detail (8).
D. HYDROGENOLYSIS OF ALCOHOLS
sAside from the possible reduction of carbonyl groups to methylene groups that may conceivably proceed through the alcohol, there are very few examples known of the hydrogenolysis of an alcohol to the methylene compound. In the case of leucopterin the hvdrogenolysis of all the hydroxyl groups does not take place, and the product is 2-amino-7-hydroxypteridine (366). The hydrogenolysis of hydroxysparteine to sparteine (112) may also be cited in this ection.
E. REDUCTION OF NITROGEN-OXYGEN UNSATURATED COMPOUNDS
1. Hydroxylamines
A nickel cathode and a basic catholyte were preferred for the reduction of aromatic hydroxylamines to szoxy compounds or to amines. Liquid ammonia has been used as a solvent in the reduction of phenylhydroxylamine to benzenediazoic acid (289).
2. Nitroso compound
The reduction of a nitrogen-nitroso compound to the corresponding hydrazine was best carried out in an acidic catholyte at cathodes of copper, lead, mercury, and cadmium, while a nickel or platinum cathode and a basic catholyte were preferred for the reduction of a carbon-nitroso compound to the corresponding amine. For example, 1,1-dimethylhydrazine was obtained in 90 per cent yield by the electroreduction of N-nitroso dimethylamine in aqueous sulfuric acid at a lead cathode (152) and 2,4,5-triamino-6-hydroxypyrimidine was obtained in 80-85 per cent yield by the reduction of 2,4-diamino-6-hydroxy-5-nitrosopyrimidin
3. Azoxy compounds
The reduction of azoxy compounds to azo, hydrazo, or amino compounds took place quite readily in an acid solution. In this manner, azoxybenzene gave azobenzene, hydrazobenzene, benzidine, and aniline (30).
4. Aliphatic nitro compounds
The reduction of aliphatic nitro compounds was carried out in an acidic catholyte at a wide variety of cathodes. Generally, the cathodes of lower overvoltage such as copper, tin, or nickel, permitted partial reduction of the hydroxylamine (which can be reduced further under certain conditions as mentioned above), whereas the cathodes of higher overvoltage, such as mercury and lead, favored the complete reduction of the nitro group to the amino group.
A number of 2-aminoethyl compounds have been prepared by the reduction of a,o-unsaturated nitro compounds at a lead cathode in hydrochloric acid.
5. Aromatic nitro compounds
The aromatic nitro group is perhaps the easiest of all unsaturated groups to reduce electrolytically. The platinum cathode has been principally used to bring about this reduction, but procedures have been described for the reduction of aromatic nitro compounds at almost all other known cathodes. The pH of the catholyte is of prime importance in controlling the product of the electroreduction of an aromatic nitro compound; in fact, several chapters of a book (42) have been devoted to the reduction of aromatic nitro compounds in basic, neutral and acidic catholytes.
Briefly, the effect of the pH of the catholyte upon the reduction of aromatic nitro compounds may be divided into four classes. In a basic catholyte bimolecular products, the azo-, azoxy-, and hydrazo-compounds, predominate. Presumably this is due to the base-catalyzed condensation of such intermediate
reduction products as the nitroso- and hydroxylamine compounds.
The reduction to azoxy compounds is general except for o- and p-nitrophenols, nitroanilines, and nitro-Nand -N,N-substituted anilines, and certain hindered nitro compounds. Generally a cathode of low hydrogen overvoltage, such as nickel, and an alkaline solution are used. The low hydrogen overvoltage prevents the azoxy compound from being reduced. The azoxy compound is insoluble in the eatholyte and precipitates out. In order to prepare the azo compound the azoxy compound must be kept in solution. Generally the addition of alcohols or salts of aromatic sulfonic acids aids this solubility. A nickel cathode is most generally used at a temperature near the boiling point of the catholyte for the formation of azo compounds. In order to form hydrazo compounds the conditions for the formation of an azo compound are followed and then the cathode current density is lowered. In a neutral or weakly acidic medium it is sometimes possible to isolate the aromatic hydroxylamine and nitroso compounds. The reduction of aromatic nitro compounds to p-aminophenols has been known for some time. At a smooth platinum cathode in sulfuric acid the reduction of aromatic hydroxylamines does not proceed rapidly; rather, in the acid medium they rearrange to the p-aminophenols. In the case of 3-nitrophthalic acid the hydroxylamine, as formed, reacts with the carbonyl group to give benzisoxazolone-4carboxylic acid (120).
In strong acid solutions amines result from the reduction of nitro compounds, while in a very strongly acidic catholyte benzidine may be obtained, probably through the acid-catalyzed benzidine rearrangement of intermediate hydrazo compounds.
An excellent procedure for the preparation of 3chlorotoluquinone has been described (48). 6-Chloro2-nitrotoluene is reduced electrolytically to the p-aminophenol, and the reaction mixture is oxidized directly to the quinone in good yield. It is often possible to reduce the nitro group without further reducing other groups present in the molecule.
F. REDUCTION OF NITROGEN-NITROGEN UNSATURATED COMPOUNDS
1. Azo compounds
Azo compounds were reduced easily to the corresponding hydrazo compounds, or cleaved to two molecules of the corresponding amines, in either basic or acidic catholytes at many different cathodes, including platinum. Azobenzene, for example, has been reduced to hydrazobenzene, benzidine, and aniline (30, 328). Coke has been found to be an efficient cathode for this reduction (328).
2. Diazonium salts
A few diazonium salts have been reduced at a number of high-overvoltage cathodes in acidic catholytes to the corresponding substituted phenyihydrazines. In this manner phenylhydrazine (269) and oand p-methoxyphenylhydrazines (101) have been obtained in good yield; in the case of the methoxy compounds the yields are better than those obtained by conventional chemical methods.
G. REDUCTION OF SULFUR-CONTAINING ORGANIC
COMPOUNDS
Reduction of the sodium or potassium salts of sulfonic acids at a lead cathode resulted in cleavage of the sulfonic acid residue from the organic molecule.
In an acid solution at a lead cathode aromatic sulfonyl chlorides may be reduced to sulfinic acids, disulfides, sulfoxides, or thiophenols, depending upon the duration of the electroreduction (340, 341).
Sodium thiosulfates are reduced to disulfides in neutral or alkaline catholytes at a platinum cathode.
Even with very long periods of electroreduction no mercaptans or thiophenols were ever isolated.
By the use of a lead or an amalgamated aluminum cathode and an acidic catholyte, thioacetamides are reduced to the corresponding amines with complete removal of the sulfur.
Reduction of nitrated aromatic thiocyanates, in which the two groups were adjacent to one another, resulted in interaction of the reduced forms to give a thiazole (104).
Reduction of disulfides in an acid solution at a lead cathode resulted in reductive cleavage of the molecule between the two sulfur groups to give the corresponding mercaptans (291). Homocystine has been converted to homocysteine by use of a mercury cathode and a basic catholyte (15). Thiolactams were reduced to the corsponding cyclic amines at a lead cathode in an acidic catholyte.
H. REDUCTION OF MISCELLANEOUS
ORGANIC COMPOUNDS
Hydrogenolysis of organic halogen compounds has been found to take place in either acidic or basic catholytes at palladium, lead, copper, mercury, and amalmated zinc cathodes. Generally better yields, in a shorter period of time, were obtainable in an acid catholyte. The reduction of 9-(o-iodophenyl)acridine to (o-iodophenyl)acridan and then to 9-phenylacridan at a mercury cathode can be performed selectively by controlling the potential (208). The removal of an aromatic halogen cannot always be predicted (see reference 13).
Arsenic compounds in an acid catholyte were reduced to arseno or arsinic type compounds; generally a high concentration of acid favored the former.
Chemistry is our Covalent Bond
(Moderator)
06-07-03 18:59
No 438549
IV. REFERENCES
(1) AIKAZYAN, E. A., AND PLISKOV, Y. V.: Zhur. Fiz. Khim. 31, 205 (1957).
(2) ALLEN, M. J.: J. Org. Chem. 15, 435 (1950).
(3) ALLEN, M. J.: Anal. Chem. 22, 804 (1950).
(4) ALLEN, M. J.: J. Am. Chem. Soc. 72, 3797 (1950). (5) ALLEN, M. J.: J. Chem. Soc. 1951, 1598. (6) ALLEN, M. J.: J. Am. Chem. Soc. 73, 3503 (1951).
(7) ALLEN, M. J.: Proc. 6th Meeting Intern. Comm. Electrochem. Thermodynam. and Kinet. 1955, 481; Chem.Abstracts 50,10574 (1956).
(8) ALLEN, M. J.: Organic Electrode Processes. Reinhold Publishing Corporation, New York (1958).
(9) ALLEN, M. J., AND COHEN, H.: J. Electrochem. Soc. 106,
451(1959).
(10) ALLEN, M. J., AND CORWIN, A. H.: J. Am. Chem. Soc. 72, 114 (1950).
(11) ALLEN, M. J., AND CORWIN, A. H.: J. Am. Chem. Soc. 72, 117 (1950).
(12) ALLEN, M. J., FEARN, J. E., AND LEVINE, H. A.: J. Chem. Soc. 1952, 2220.
(13) ALLEN, M. J., AND OCAMPO, J.: J. Electrochem. Soc. 103,
452, 682 (1956).
(14) ALLEN, M. J., SINAGUSA, J. A., AND PIERSON, W.: J. Chem. Soc. 1960, 1045.
(15) ALLEN, M. J., AND STEINMAN, H. G.: J. Am. Chem. Soc.
74, 3932 (1952).
(16) ANTROPOV, L. I.: Zhur. Fiz. Khim. 24, 1428 (1950).
(17) ANTROPOV, L. I., AND VAGRAMYAN, N. T.: Zhur. Fiz. Khim. 25, 409 (1951).
(18) ANZIANI, P., AUBRY, A., AND CORNUBERT, R.: Compt. rend. 225, 878 (1947).
(19) ATANASIU, I., BLUM, L., AND POPA, M.: Bull. inst. politehnic Bucure,§ti 20, No. 2, 103 (1958); Chem. Abstracts 54,3002 (1960).
(20) ATANASIU, I., AND DUMITRU, Gh.: Rev. chim. (Bucharest) 9,129 (1958); Chem. Abstracts 53, 5916 (1959).
(21) ARAI, T., AND OGURI, T.: Bull. Chem. Soc. Japan 33, 1018 (1960).
(22) Atlas Powder Company: British patents 533,884 (1941) and 533,885 (1942); Chem. Abstracts 36, 975 (1942). (23) BAEZNER, C., AND GARDIOL, H.: Ber. 39, 2512 (1906).
(24) BAHR, T.: Ges. Abhandl. Kenntnis Kohle 11, 246 (1934); Chem. Abstracts 29, 6512 (1935).
(25) BANCROFT, W. B., AND GEORGE, A. B.: Trans. Am. Electrochem. Soc. 57, 399 (1930).
(26) BELEN'KAYA, N. G., AND BELOZERSKII, N. A.: Zhur. Obshchel Khim. 19, 1664 (1949); Chem. Abstracts 44, 956 (1950).
(27) BEREZOVSKII, V. M., AND VARKOV, V. S.: Zhur. Obshchel Khim. 23, 100 (1953); Chem. Abstracts 47, 5821 (1953). (28) BEREZOVSKIi, V. M., AND SOBOLEV, Y. P.: Zhur. Obshchel Khim. 28, 261 (1958).
(29) BEREZOVSKII, V. M., AND SOBOLEV, Y. P.: Khim. Nauka i. Prom. 3, 677 (1958); Chem. Abstracts 53, 3943 (1959). (30) BERGMAN, I., AND JAMES, J. C.: Trans. Faraday Soc. 50, 60 (1954).
(31) BILLITER, J.: Die technische Elektrolyse der Nichtmetalle. III. Elektrolytische Oxydationen and Reduktionen. Springer Verlag, Berlin (1954).
(32) BILLITER, J.: Z. Elektrochem. 7, 683, 959 (1901).
(33) BIRCHER, L. J., AND BRUBAKER, M. M.: Organic Syntheses, Collective Vol. I, p. 485. John Wiley and Sons, Inc., New York (1941).
(34) BLADON, P., CORNFORTH, J. W., AND JAEGER, R. H.: J. Chem. Soc. 1958, 863.
(35) BLICKE, F. F., DOORENBOS, N. J., AND Cox, R. H.: J. Am. Chem. Soc. 74, 2924 (1952).
(36) BLUDWORTH, J. E., ROLUSON, M. D., AND TRUBY, H. A.: U.S. patent 2,462,301 (1949); Chem. Abstracts 43, 3300 (1949).
(37) BOCKRIS, J.O'M., AND CONWAY, B. E.: Modern Aspects of Electrochemistry. 1. Electrode Kinetics, pp. 180-276. Academic Press Inc., New York (1954).
(38) BRESSE, J. C.: Thesis, University of Illinois (1945); quoted in reference 323, p. 427.
(39) BREWSTER, J. H.: J. Am. Chem. Soc. 80, 6284 (1958).
(40) BRINTZINGER, H., AND EGGERS, V.: Z. Elektrochem. 56, 158 (1952).
(41) BRINTZINGER, H., ZIEGLER, H. W., AND SCHNEIDER, E.: Z. Elektrochem. 53,109 (1949).
(42) BROCKMAN, C. J.: Electro-organic Chemistry, John Wiley and Sons, Inc., New York (1926).
(43) BROWN, K. R.: U.S. patent 2,280,887 (1942); Chem. Abstracts 36, 5433 (1942).
(44) BROWN, K. R.: Canadian patent 402,883 (1942); Chem. Abstracts 36, 2481 (1942).
(45) BRUCKNER, V., KOVeACS, J., AND Kovics, K.: Acta Univ.
Szedediensis Acta Chem. et Phys. (N.S.] 2, 18 (1948). (46) BRUHUR, V., KovAcs, J., AND Kovkcs, K.: Ber. 77, 610 (1944).
(47) CAMPBELL, K. N., AND YOUNG, E. E.: J. Am. Chem. Soc. 65, 965 (1943).
(48) CASON, J., ALLEN, C. F., AND GOODWIN, S.: J. Org. Chem. 13, 403 (1948). CASPARI, W.: Z. physik. Chem. 30, 89 (1899). CHAMBERS, T. S., AND SLOTTERBECK, 0. C.: U.S. Patent 2,485,258 (1949); Chem. Abstracts 44, 4807 (1950).
(51) CHILESOTTI, A.: Z. Elektrochem. 7, 768 (1900).
(52) CLAUSON-KAAS, N. K. F. W., AND LIMBORG, F.: U.S. patent 2,714,576 (1955); Chem. Abstracts 49, 15573 (1955).
(53) COLEMAN, G. H., AND JOHNSON, H. L.: Organic Syntheses, Collective Vol. III, p. 60. John Wiley and Sons, Inc., New York (1955).
(54) CONDIT, P. C.: U.S. patent 2,537,304 (1951); Chem. Abstracts 45, 2341 (1951).
(55) CREIGHTON, H. J.: Can. Chem. Proc. Ind. 26, 690 (1942); Chem. Abstracts 37, 1088 (1943).
(56) CREIGHTON, H. J.: J. Electrochem. Soc. 99, 127C-129C (1952).
CREIGHTON, H. J., AND HALES, R. A.: U.S. patent 2,458,895 (1949); Chem. Abstracts 43, 2104 (1949).
(58) CREIGHTON, H. J., AND KOEHLER, W. A.: Principles and Practice of Electrochemistry. Vol. II. John Wiley and Sons, Inc., New York (1944). DELAHAY, P.: Ann. Rev. Phys. Chem. 8, 229 (1957).
DE STEVENS, G., AND WERNER, L. H.: Swiss patent 337,205 (1959); Chem. Abstracts 54, 21148 (1960).
(61) DEY, B. B., GOVINDACHARI, T. R., AND RAJAGOPALAN, S. C.: J. Sci. Ind. Research (India) 4, 559 (1946).
(62) DEY, B. B., GOVINDACHARI, T. R., AND RAJAGOPALAN, S. C.: J. Sci. Ind. Research (India) 4, 574 (1946).
(63) DEY, B. B., GOVINDACHARI, T. R., AND RAJAGOPALAN, S. C.: J. Sci. Ind. Research (India) 4, 637 (1946).
(64) DEY, B. B., GOVINDACHARI, T. R., AND RAJAGOPALAN, S. C.: J. Sci. Ind. Research (India) 4, 645 (1946).
(65) DEY, B. B., GOVINDACHARI, T. R., AND RAJAGOPALAN, S. C.: J. Sci. Ind. Research (India) 5, 75 (1946). DEY, B. B., GOVINDACHARI, T. R., AND RAJAGOPALAN, S. C.: Indian patent 34,757 (1948); Chem. Abstracts 44, 6886 (1950). DEY, B. B., GOVINDACHARI, T. R., AND UDUPA, H. V.: Current Sci. 15, 163 (1946).
(68) DEY, B. B., MALLER, R. K., AND PAT, B. R.: J. Sci. Ind. Research (India) 7, 107 (1948).
(69) DEY, B. B., MALLER, R. K., AND PAT, B. R.: J. Sci. Ind. Research (India) 7, 113 (1948).
DEY, B. B., MALLER, R. K., AND PAT, B. R.: J. Sci: Ind. Research (India) 7, 198 (1948). DEY, B. B., MALLER, R. K., AND PAT, B. R.: J. Sci. Research (India) 8, 206 (1949). DEY, B. B., MALLER, R. K., AND PAT, B. R.: J. Sci. Ind. Research (India) 9, 55 (1950). DEY, B. B., MALLER, R. K., PAT, B. R., AND UDUPA, H. V.: Indian patent 39,427 (1950); Chem. Abstracts 44, 9279 (1950).
(74) DEY, B. B., MALLER, R. K., AND PAT, B. R.: Indian patent 39,429 (1950); Chem. Abstracts 44, 9278 (1950).
(75) DEY, B. B., MALLER, R. K., AND PAT, B. R.: J. Sci. Ind. Research (India) 10, 175 (1951).
(76) DEY, B. B., PAT, B. R., AND MALLER, R. K.: J. Sci. Ind. Research (India) 7, 71 (1948).
(77) DEY, B. B., AND UDUPA, H. V.: J. Sci. Ind. Research (India) 6, 83 (1947).
(78) DEY, B. B., UDUPA, H. V., AND PAT, B. R.: Current Sci.
16,186 (1947).
(79) DEY, B. B., AND UDUPA, H. V.: Current Sci. 22, 371 (1953).
(80) DIAMOND, G. B., AND SOFFER, M. D.: J. Am. Chem. Soc. 74, 4126 (1952).
(81) DICKENSON, H. G., AND WEISS, J.: British patent 629,042 (1949); Chem. Abstracts 44, 1836 (1950).
(82) Dillon, C. S.: British patent 611,674 (1948); Chem. Ab
stracts 43, 4589 (1949).
(83) Dluzniewski, A.: Dissertationes Pharm. 11, 165 (1959); Chem. Abstracts 54, 12834 (1960).
(84) Doolittle, A. K.: Trans. Am. Electrochem. Soc. 45, 57 (1924).
(85) DOLLIVER, M. A., AND SEMENOFF, S.: U.S. patent 2,717,236 (1955); Chem. Abstracts 49, 15573 (1955). DUNET, A., ROLLET, J., AND WILLEMART, A.: Bull. soc. chim. France 1950, 877.
(87) DUNET, A., AND WILLEMART, A.: Compt. rend. 222, 1443 (1946). DUNET, A., AND WILLEMART, A.: Bull. soc. chim. France 1948, 887.
(89) ELBS, K.: J. prakt. Chem. [2] 43, 39 (1891).
(90) ELBS, K.: Z. Elektrochem. 3, 48 (1896).
(91) ELBS, K.: Ber. 38, 4012 (1905).
(92) ELOFSON, R. M.: J. Org. Chem. 25, 305 (1960).
(93) ELVING, P. J., ROSENTHAL, I., AND MARTIN, A. J.: J. Am.Chem. Soc. 77, 5218 (1955).
(94) ERSHOV, B. P., AND ZEPALOVA-MIKHAILOVA, L. A: J. Applied Chem. (U.S.S.R.) 16, 383 (1943); Chem. Abstracts 38, 6212 (1944).
(95) FARBENFABRIKEN VORM. F. BAYER & Co.: German patent 310,023 (1916); Chem. Abstracts 15, 2038 (1921).
(96) FARNAU, E. F.: J. Phys. Chem. 16, 249 (1912); Chem. Abstracts 6, 1612 (1912).
(97) FAVORSKAYA, I. A.: Zhur. Obshchel Khim. 18, 52 (1948).
(98) FERLES, M.: Chem. Listy 52, 668 (1958); Chem. Abstracts 52, 13724 (1958).
(99) FERLES, M.: Chem. Listy 52, 674 (1958); Chem. Abstracts 52, 13724 (1958).
(100) FESTER, G., AND SCHIVAZAPPA, M.: Z. anorg. allgem. Chem. 171, 163 (1928).
Chemistry is our Covalent Bond
(Moderator)
06-07-03 19:06
No 438550
(101) FIOSHIN, M. Y., GIRINA, G. P., AND MAMAEV, V. P.: Zhur. Obshchel Khim. 26, 2311 (1956).
(102) FICHTER, F.: Ber. 47, 2015 (1914).
(103) FICHTER, F.: Organische Elektrochemie. Edwards Brothers, Ann Arbor, Michigan (1946).
(104) FICHTER, F., AND BECK, T.: Ber. 44,3656 (1911).
(105) FTERZ-DAVID, H. E., BLANGEY, L., AND UHLIG, M.: Helv. Chim. Acta 32, 1414 (1949).
(106) FLORIANOVICH, G. M.: Zhur. Fiz. Khim. 31, 626 (1957). (107) FOSTER, D. G.: J. Chem. Educ. 28,626 (1951).
(108) FRANCE, W. G., AND TURK, A.: J. Phys. & Colloid Chem. 53, 482 (1949).
(109) FREAS, R.: Trans. Am. Electrochem. Soc. 40,109 (1921). (110) FUTAKE, Z.: Japanese patent 174,272 (1946); Chem. Abstracts 44, 961 (1950).
(111) GAKENHEIMER, W. C., AND HARTUNG, W. H.: J. Org. Chem. 9, 85 (1944).
(112) GALINORSKY, F. G., AND SCHMID, H.: Monatsh. Chem. 79, 322 (1948).
(113) GALINORSKY, F., AND REICHARD, A.: Ber. 77,138 (1944).
(114) GEVER, G., O'KEEFE, C., DRAKE, G., EBETINO, F.,MICHELS, J., AND HAYES, K.: J. Am. Chem. Soc. 77, 2277 (1955).
(115) GLASSTONE, S.: Trans. Faraday Soc. 19, 574 (1924).
(116) GLASSTONE, S.: Ind. Chemist 5, 423 (1929).
(117) GLASSTONE, S.: Ind. Chemist 6, 201 (1930).(49) (50)(86)(88)(57)(59) (60)(66) (67)(70)
(72) (73)
(118) GLASSTONE, S.: Textbook of Physical Chemistry, D. Van Nostrand Company, Inc., New York (1940).
(119) GLASSTONE, S., AND HICKLING, A.: Electrolytic Oxidation and Reduction. D. Van Nostrand Company, Inc., New York (1956).
(120) GLEN, K., AND PFANNSTEIL, K.: J. prakt. Chem. 146, 129 (1936).
(121) GOLOVCHANSKAYA, A. P.: J. Gen. Chem. (U.S.S.R.) 10, 435 (1940).
(122) GOLOVCHANSKAYA, A. P.: J. Gen. Chem. (U.S.S.R.) 11, 608 (1941).
(123) GoODINGs, E. P., AND WILSON, C. L.: Trans. Electrochem. Soc. 88, 77 (1945).
(124) GRABETSKII, A. A., AND LEVCHENKO, V. V.: J. Gen. Chem.(U.S.S.R.) 17, 1564 (1947).
(125) GRAHAME, D. C.: Ann. Rev. Chem. 3, 247 (1952).
(126) GRUBINA, L. M., AND STENDER, V. V.: J. Applied Chem.(U.S.S.R.) 13, 1028 (1940).
(127) GUNTHER, G., AND STANDE, H.: Z. Elektrochem. 56, 673(1952).
(128) GYka, R. G. V.: Hungarian patent 124,476 (1940); Chem. Abstracts 35, 4921 (1941).
(129) HABER, F.: Z. Elektrochem. 4, 510 (1897).
(130) HABER, F.: Z. physik. Chem. 32, 193 (1900).
(131) HABERLAND, H.: German patent 890,643 (1953); Chem.Abstracts 50, 12100 (1956).
(132) HALES, R. A.: U.S. patent 2,289,189 (1943); Chem.Abstracts 37, 42 (1943).
(133) HALES, R. A.: U.S. patent 2,300,218 (1943); Chem. Abstracts 37, 1660 (1943). (134) HALES, R. A.: U.S. patent Abstracts 37, 2277 (1943).
(135) HARMON, R. E., AND CASON, J.: J. Org. Chem. 17, 1047 (1952).
(136) HARMON, R. E., AND CASON, J.: J. Org. Chem. 17, 1058 (1952).
(137) HAEUSSERMANN, C.: Chem. Ztg. 17, 129, 209 (1893).
(138) HAYASHI, S., SUGINO, K., AND MIZUGUCHI, J.: Japanese patent 176,226 (1948); Chem. Abstracts 45, 5048 (1951).
(139) HEISEL, P.: German patent 848,807 (1952); Chem. Abstracts 50, 6229 (1956).
(140) HEMPTINNE, X. DE, AND JUNGENS, J. C.: Z. physik. Chem. 15, 137 (1958).
(141) HENNIG, G. H., AND KIMBALL, G. H.: J. Phys. Chem. 12, 415 (1944).
(142) HEPTI, H. R., AND KOLB, W.: U.S. patent 2,507,973
(1950); Chem. Abstracts 45, 2340 (1951).
(143) HEY, P.: Quart. J. Pharm. Pharmacol. 20,129 (1947).
(144) HIRAYAMA, H.: J. Chem. Soc. Japan 66, 15 (1945); Chem. Abstracts 42, 7169 (1948).
(145) HIRAYAMA, H.: Ann. Repts. Shionogi Research Lab. (Osaka) 1953, 41.
(146) HIRAYAMA, H., AND KUBOTA, T.: Japanese patent 2671 (1951); Chem. Abstracts 47, 426 (1953).
(147) HOBDAY, G. I., AND SHORT, W. F.: J. Chem. Soc. 1943, 609. 148) HOFFMANN-LA ROCHE AND Co., A.-G.: Swiss patent 258, 581 (1949); Chem. Abstracts 44, 4352 (1950).
(149) HOFFMANN, W.: Dissertation, University of Giessen (1914); quoted on p. 511 of reference 323.
(150) HoLLECK, L., AND SCHMIDT, H.: Z. Elektrochem. 59, 56 (1955).
(151) HONDA, K., YOKOUCHI, R., AND KIKUCHI, S.: J. Electrothem. Soc. Japan 20, 15 (1952); Chem. Abstracts 46, 4930 (1952).
(152) HORVITZ, D., AND CERWONKA, E.: U.S. patent 2,916,426 (1959); Chem. Abstracts 54, 6370 (1960).
(153) HoSHINO, T., AND SATO, T.: Japanese patent 4359 (1950); Chem. Abstracts 47, 3341 (1953).
(154) INGERSOLL, A. W.: Organic Syntheses, Collective Vol. I, 2nd edition, p. 311. John Wiley and Sons, Inc., New York (1941).
(155) INGERSOLL, A. W., BIRCHER, L. J., AND BRUBAKER, M. M.: Organic Syntheses, Collective Vol. I, p. 485. John Wiley and Sons, Inc., New York (1941).
(156) ISHIFUKU, K., SAKURAI, H., AND OKAMOTO, H.: Japanese patent 180,563 (1949); Chem. Abstracts 46, 3432 (1952).
(157) ISHIWATA, S., AND NozAKI, T.: J. Pharm. Soc. Japan 71, 1257 (1951).
(158) ITO, A.: J. Soc. Org. Synthetic Chem. (Japan) 11, 252 (1953).
(159) IZGARYSHEV, N. A., AND ARYAMOVA, I. I.: Zhur. Obshchel Khim. 18, 337 (1948).
(160) IZGARYSHEV, N. A., AND ARYAMOVA, I. I.: Doklady Akad. Nauk S.S.S.R. 84, 313 (1952).
(161) IZGARYSHEV, N. A., AND FIOSHIN, M. Y.: Doklady Akad. Nauk S.S.S.R. 90,189 (1953).
(162) IZGARYSHEV, N. A., AND PETROVA, A. A.: Zhur. Fiz. Khim. 24, 745 (1950).
(163) KABOZEV, N. I., AND MONBLANOVA, V. V.: J. Phys. Chem. Russia 7, 645 (1936); Chem. Abstracts 30, 8040 (1936). (164) KAUFLER, F.: Z. Elektrochem. 13, 633 (1907). (165) KAUFMAN, F., COOK, H. J., AND DAVIS, S. M.: J. Am. Chem. Soc. 74, 4997 (1952).
(166) KAWAMURA, F., AND SUZULI, S.: J. Chem. Soc. Japan, Ind. Chem. Sect. 55, 476 (1952); Chem. Abstracts 48, 3167 (1954).
(167) KE, B.: J. Am. Chem. Soc. 78, 3649 (1956).
(168) KHOMYAKOV, V. G., FIOSHIN, M. Y., AND KRUGLIKOV,
S. S.: Khim. Nauka i. Prom. 3, 432 (1958).
(169) KIKUCHI, S., AND UTSUNOMIYA, T.: J. Chem. Soc. Japan, Ind. Chem. Sect. 54, 534 (1951); Chem. Abstracts 47, 6277 (1953).
(170) KIKUCHI, S., AND MASAGO, H.: J. Soc. Sci. Phot. Japan 14, 11 (1951); Chem. Abstracts 46, 7448 (1952).
(171) KINDLER, K.: Ber. 57, 773 (1924).
(172) KNUNYANTS, I. L., AND VYAZANKIN, N. S.: Doklady Akad. Nauk S.S.S.R. 113, 112 (1957).
(173) KODAMA, K.: J. Pharm. Soc. Japan 63, 54 (1943); Chem. Abstracts 45, 5169 (1951).
(174) KONDO, Y.: Japanese patent 1830 (1954); Chem. Abstracts 49, 82 (1955).
(175) KOPERINA, A. V., AND GAVRILOV, N. I.: J. Gen. Chem. (U.S.S.R.) 17, 1651 (1947).
(176) KOPERINA, A. V., AND KLYUCHAREVA, M. M.: J. Gen. Chem. (U.S.S.R.) 11, 51 (1941).
(177) Kovkcs, J.: Acta Univ. Szegediensis Acts, Chem. et Phys. [N.S.] 1, 109 (1943); Chem. Abstracts 42, 173 (1948).
(178) KovAcs, J.: Acta Univ. Szegediensis Chem. et Phys. 2, 56 (1948); Chem. Abstracts 44, 6384 (1950).
(179) KRAFT, M. Y., KORZINA, 0. I., AND MovozovA, A. S.: Sbornik Statei Obshchel Khim. 2, 1356 (1953); Chem. Abstracts 49, 5347 (1955).
(180) KRAMLI, A., AND VARGHA, L.: U.S. patent 2,356,596 (1944); Chem. Abstracts 39, 29 (1945).
(181) KuKoz, F. I., AND ANTROPOV, L. I.: Zhur. Fiz. Khim. 32, 2294 (1958).(182) KUWATA, T.: Japanese patent 9966 (1958); Chem. Abstracts 54, 5298 (1960).
(183) LATIMER, W. M., AND HILDEBRAND, J. H.: Reference Book of Inorganic Chemistry, The Macmillan Company, New York (1940).
(184) LAW, H. D.: J. Chem. Soc. 89, 1520 (1906). 2,303,210 (1943); Chem. 38
(185) LAW, H. D.: J. Chem. Soc. 99,1113 (1911).
(186) LAW, H. D.: J. Chem. Soc. 101, 1016 (1912). (187) LAW, H. D.: J. Chem. Soc. 101, 1544 (1912).
(188) LEBEDEVA, A. I.: Compt. rend. acad. sci. U.R.S.S. 42, 70; Doklady Akad. Nauk S.S.S.R. 42, 71(1944).
(189) LEBEDEVA, A. I.: Zhur. Obshchel Khim. 18, 1161 (1948).
(190) LEBEDEVA, A. I., AND MISHNINA, T. A.: J. Gen. Chem. (U.S.S.R.) 21, 1227 (1951) (Engl. translation).
(191) LEBEDEVA, A. I., AND MISHNINA, T. A.: Zhur. Obshchel Khim. 25, 1507 (1955).
(192) LEEDS, M. W., AND SMITH, G. B. L.: J. Electrochem. Soc. 98, 129 (1951).
(193) LEONARD, N. J., SWANN, S., JR., AND DRYDEN, H. L.: J. Am. Chem. Soc. 74, 2871(1952).
(194) LEONARD, N. J., SWANN, S., JR., AND FIGUERAS, J., JR.J. Am. Chem. Soc. 74, 4620 (1952).
(195) LEONARD, N. J., SWANN, S., JR., AND FULLER, G.: J. Am. Chem. Soc. 76, 3193 (1954).
(196) LEONARD, N. J., SWANN, S., JR., AND FULLER, G.: J. Am. Chem. Soc. 75, 5127 (1953).
(197) LEONARD, N. J., SWANN, S., JR., AND MOTTUS, E. H.: J. Am. Chem. Soc. 74, 6251(1952).
(198) LESLIE, W. M., AND BUTLER, J. A. V.: Trans. Faraday Soc. 32, 989 (1936).
(199) LEUCHS, H., AND WEGENER, W.: Ber. 63, 2215 (1930). (200) LEVCHENKO, V. V.: Zhur. Obshchel Khim. 11, 686 (1941).
(201) LEVCHENKO, V. V.: Zhur. Obshchel Khim. 17, 1656 (1947).
(202) LEVCHENKO, V. V.: Zhur. Obshchel Khim. 18, 1237 (1948).
(203) LEVCHENKO, V. V., AND MALESHKO, K. V.: Zhur. Obshchel Khim. 20, 831 (1950).
(204) LEVCHENKO, V. V., AND ZATS, A. A.: Zhur. Obshchel Khim. 22, 1253 (1952).
(205) LEVINE, H. A., AND ALLEN, M. J.: J. Chem. Soc. 1952, 254.
(206) LEWIS, R. W., AND BROWN, 0. W.: Trans. Electrochem. Soc. 84, 135 (1943).
(207) LINGANE, J. J.: Anal. Chem. 22, 1169 (1950).
(208) LINGANE, J. J., SWAIN, C. G., AND FIELDS, M.: J. Am. Chem. Soc. 65, 1348 (1943).
(209) LISTOPAnov, V. V., AND ANTROPOV, L. I.: Nauch. Trudy Novocherkassk. Politekh. Inst. 34, 87 (1956); Chem. Abstracts 53, 13836 (1959).
(210) LOB, W., AND MOORE, R. W.: Z. physik. chem. 47, 418 (1904).
(211) LIEBEN, A.: Monatsh. Chem. 18, 582 (1897).
(212) LUND, H.: Acta Chem. Scand. 11, 283 (1957).
(213) MCGTINE, T. H., AND DULL, M. F.: J. Am. Chem. Soc. 69,1469 (1947).
(214) McKEE, R. H., AND BROCKMAN, C. J.: Trans. Electrochem. Soc. 62, 203 (1932).
(215) MCKEE, R. H., AND GERAPOSTOLOU, B. G.: Trans. Electrochem. Soc. 68, 329 (1935).
(216) MCMILLAN, G. W.: U.S. patent 2,485,982 (1949); Chem. Abstracts 44, 1836 (1950).
(217) MADAUS, J. H., AND URBACH, H. B.: U.S. patent 2,918,418 (1959); Chem. Abstracts 54, 11774 (1960).
(218) MANDELL, L., POWERS, R. M., AND DAY, R. A.: J. Am. Chem. Soc. 80, 5284 (1958).
(219) MARTINYUK, G. A., AND SHLYGIN, A. I.: Zhur. Fiz. Khim. 32, 1907 (1958).
(220) MASU1, M., SAYO, H., AND NOMURA, Y.: Pharm. Bull. 4, 337 (1956).
(221) MASUNO, M., ASAHARA, T., KUROIWA, S., SHIMIZU, K., AND NAKANO, J.: J. Chem. Soc. Japan, Ind. Chem. Sect. 52, 151 (1949); Chem. Abstracts 45, 1884 (1951).
(222) MAY, J. A., AND KOBE, K. A.: J. Electrochem. Soc. 97, 183 (1950).
(223) METTLER, C.: Ber. 37, 3692 (1904).
(224) METTLER, C.: Ber. 39, 2933 (1906).
(225) MIZUGUCHI, J., AND MATSUMOTO, S.: Yakugaku Zasshi 78, 129 (1958); Chem. Abstracts 52, 8794 (1958).
(226) MoMOSE, T.: Japanese patent 3073 (1951); Chem. Abstracts 47, 5439 (1953).
(227) MUHLEMANN, H., AND URWYLER, H.: Pharm. Acta Helv. 26, 181 (1951).
(228) MULLER, E.: Z. Elektrochem. 16, 236 (1910). (229) NERNST, W.: Z. Elektrochem. 7, 267 (1900).
(230) NORRIS, J. F., AND CUMMINGS, E. 0.: Ind. Eng. Chem. 17, 305 (1925).
(231) OCHIAI, E., AND KATAOKA, H.: J. Pharm. Soc. Japan 62,
241 (1942); Chem. Abstracts 45, 5150 (1951).
(232) OCHIAI, E., TESHIGAWARA, T., AND NAITO, T.: J. Pharm. Soc. Japan 65, 429 (1945); Chem. Abstracts 45, 8376 (1951).
(233) ODO, K., AND SUGINO, K.: J. Electrochem. Soc. 104, 160 (1957).
(234) OGANESYAN, A. S., AND ANTROPOV, L. I.: Doklady Akad. Nauk Armyan. S.S.R. 21, 81 (1955); Chem. Abstracts 50, 1496 (1956).
(235) OGURA, K.: Mem. Coll. Sci. Kyoto Imp. Univ. 12A, 339 (1929); Chem. Abstracts 24, 2060 (1930).
(236) OHKI, S.: J. Pharm. Soc. Japan 70, 92 (1950); Chem.Abstracts 44, 5867 (1950).
(237) OHKI, S., AND NOIKE, Y.: J. Pharm. Soc. Japan 72, 490 (1952); Chem. Abstracts 47, 6418 (1953).
(238) OHTA, M.: J. Chem. Soc. Japan 63, 1762 (1942); Bull. Chem. Soc. Japan 17, 485 (1942); Chem. Abstracts 41, 3752 (1947).
(239) OISHI, Y.: J. Chem. Soc. Japan, Ind. Chem. Sect. 56, 545 (1953); Chem. Abstracts 48, 11960 (1954).
(240) OHDAKE, R., KoJIMA, Y., AND KUSAKABE, H.: Repts. Sci. Research Inst. (Japan) 28, 199, 316 (1952); Chem. Abstracts 47, 2432, 7348 (1953).
(241) OKUDA, H., AND SAJI, K.: Science and Ind. (Japan) 28, 314 (1954); Chem. Abstracts 49, 12990 (1955).
(242) ONO, S.: J. Chem. Soc. Japan, Pure Chem. Sect. 73, 852
(1952); Chem. Abstracts 47, 11046 (1953).
(243) ONO, S.: J. Chem. Soc. Japan, Pure Chem. Sect. 74, 395 (1953); Chem. Abstracts 48, 9988 (1954).
(244) ONO, S.: Bull. Naniwa Univ. 2A, 117 (1954); Chem. Abstracts 48, 13484 (1954).
(245) ONO, S.: Bull. Naniwa Univ. 2A, 123 (1954); Chem. Abstracts 48, 13486 (1954).
(246) ONO, S.: Nippon Kagaku Zasshi 75, 1195 (1954); Chem. Abstracts 51, 12704 (1957).
(247) ONO, S. J.: J. Electrochem. Soc. Japan 23, 117 (1955); Chem. Abstracts 49, 9410 (1955).
(248) ONO, S.: Nippon Kagaku Zasshi 76, 631 (1955); Chem. Abstracts 51, 17525 (1957).
(249) ONO, S., AND HAYASHI, T.: Bull. Chem. Soc. Japan 26,11 (1953).
Chemistry is our Covalent Bond
(Moderator)
06-07-03 19:08
No 438551
(250) ONO, S., AND HAYASHI, T.: Bull. Chem. Soc. Japan 26, 232 (1953).
(251) ONO, S., AND YAMAUCHI, T.: Bull. Chem. Soc. Japan 25, 404 (1952).
(252) OROSHNIK, W., AND SPOERRI, P. E.: J. Am. Chem. Soc. 63, 338 (1941).
(253) Osipov, 0. A., AND PETRENKO, A. T.: Uchenye Zapiski Rostov.-na-Donu Univ. 25, 65 (1955); Chem. Abstracts 52, 19601 (1958).
(254) PAISS, Y., AND STEIN, G.: J. Chem. Soc. 1958, 2905.
(255) PARKER, E. A., AND SWANN, S., JR: Trans. Electrochem.
Soc. 92, 343 (1947).
(256) PASTERNAK, R.: Helv. Chim. Acta 31, 753 (1948) ELECTROLYTIC REDUCTION OF ORGANIC COMPOUNDS
39
(257) PEARL, I. A.: J. Am. Chem. Soc. 74, 4260 (1952).
(258) PoMILIo, V.: Z. Elektrochem. 21, 444 (1915).
(259) POWERS, R. M., AND DAY, R. A.: J. Org. Chem. 24, 722 (1959).
(260) PROUDFIT, C. W., AND FRANCE, W. G.: J. Phys. Chem. 46, 42 (1942).
(261) QUESNEL, G. L., SIMON, P., AND CHALAUST, R.: French patent 1,167,480 (1958); Chem. Abstracts 54, 24036 (1960).
(262) Ravenscroft, P. H., Lewis, R. W., and Brown, 0. W.: Trans. Electrochem. Soc. 84, 145 (1943).
(263) READ, H. J.: Trans. Electrochem. Soc. 80,133 (1941). (264) REGNA, P. P.: J. Am. Chem. Soc. 69, 246 (1947).
(265) Reitzenstein, F., and Runge, 0.: J. prakt. chem. [2] 71, (1905).
(266) ROSENTHAL, I., HAYES, J. R., MARTIN, A. J., AND ELVING,~P. J.: J. Am. Chem. Soc. 80, 3050 (1958).
(267) ROYER, M. E.: Compt. rend. 69,1374 (1869). (268) ROYER, M. E.: Compt. rend. 70, 731 (1870).
(269) RUETSCHI, P., AND TRUMPLER, G.: Helv. Chim. Acts 36,
1649 (1953).
(270) SAKURAI, B.: J. Shinshu Univ. 1, 1 (1951); Chem. Abstracts 48, 12580 (1954).
(271) SAKURAI, B.: Shinshu Daigaku Bunriga Kubu Kiyo No. 5, Pt. II, 11 (1955); Chem. Abstracts 54, 22096 (1960). (272) SAKURAI, B., AND ARAI, T.: Bull. Chem. Soc. Japan 28, 93 (1955).
(273) SATO, T.: Bull. Tokyo Inst. Technol. 13, 133 (1948); Chem. Abstracts 44, 10548 (1950).
(274) SATO, T.: J. Chem. Soc. Japan, Pure Chem. Sect. 71, 194 (1950).
(275) SATO, T.: J. Chem. Soc. Japan, Pure Chem. Sect. 71, 310 (1950).
(276) SAWA, S.: Japanese patent 5019 (1951); Chem. Abstracts 47, 2617 (1953).
(277) SCHOLL, C., AND KIRST, W.: Z. Elektrochem. 29, 537 (1923).
(278) SCHWABE, K.: German patent 727,739 (1942); Chem. Abstracts 37, 6566 (1943).
(279) SE, H.: Japanese patent 180,486 (1949); Chem. Abstracts 46, 2432 (1952).
(280) SEKINE, T.: J. Chem. Soc. Japan, Pure Chem. Sect. 77, 67 (1956).
(281) SEKINE, T.: K6gy6 Kagaku Zasshi 60, 918 (1957); Chem. Abstracts 53, 8883 (1959).
(282) SEKINE, T., AND SUGINO, K.: J. Electrochem. Soc. Japan 21, 383 (1953); Chem. Abstracts 48, 13485 (1954).
(283) SHASHKINA, A. V.: Vestnik Moskov, Univ., Ser. Mat., Mekh., Astron., Fiz., Khim. 13 135 (1958); Chem. Abstracts 53, 9848 (1959).
(284) SHASHKINA, A. V.: Vestnik Moskov. Univ., Ser. Mat., Mekh., Astron., Fiz. Khim. 14, 135 (1959); Chem. Abstracts 54, 11759 (1960).
(285) SHIKHIEV, I. A.: Zhur. Obshchel Khim. 20, 839 (1950).
(286) SHIKHIEv, I. A.: Dokiady Akad. Nauk Azerbaldzhan. S.S.R. 11, No. 7, 459 (1955); Chem. Abstracts 50, 11937 (1956).
(287) SHIMA, G.: Mem. Coll. Sci. Kyoto Imp. Univ. 11, 1 (1928);
Chem. Abstracts 22, 3884 (1928).
(288) SHREVE, R. N., AND CARTER, R. P.: Ind. Eng. Chem. 36, 423 (1944).
(289) SIBA, H., INouR, T., AND MIYASAKA, R.: Sci. Inst. Papers Phys. Chem. Research (Tokyo) 35, 455 (1939); Chem. Abstracts 33, 6170 (1939).
(290) SITARAMAN, M. V., AND RAMAN, V. V.: Current Sci. 16, 23 (1947).
(291) SJOBERG, B.: Ber. 75, 26 (1942).
(292) SL6TTA, K. H., AND KETHUR, R.: German patent 680,273
(1939); Chem. Abstracts 36, 1854 (1942).
(293) SLOTTA, K. H., AND SZYSZKA, G.: J. prakt. Chem. 137, 339 (1933).
(294) SLOTTERBECK, 0. C.: Trans. Electrochem. Soc. 92, 377 (1947).
(295) SMIALOWSKI, M., AND JARMOLOWICZ, H.: Bull. acad. polon. sci., Classe III, 3, 107 (1955); Chem. Abstracts 49, 12990 (1955).
(296) SMIRNOVA, M. G., SMIRNOV, V. A., AND ANTROPOV, L. I.:Trudy Novocherkassk. Politekh. Inst. im S. Ordzhonikidze 79, 43 (1959); Chem. Abstracts 54, 24011 (1960).SMITH, G., AND LEEDS, M. W.: U.S. patent 2,589,635 (1952); Chem. Abstracts 46, 4937 (1952).
(298) SOCIfTA ANON. POUR L'INDUSTRIE CHIMIQUE A BALE: Swiss patent 73,098 (1916); Chem. Abstracts 11, 757(1917).
(299) SocIATA ANON. POUR L'INDUSTRIE CHIMIQUE A BALE:Japanese patent 31,757 (1917); Chem. Abstracts 12,2168 (1918).
(300) SOLANKI, D. N.: Trans. Electrochem. Soc. 88, 97 (1945). (301) SORM, F., AND ARIENT, J.: Collection Czechoslov. Chem. Communs. 15, 175 (1950).
(302) SPRETER, V. C., AND BRINER, E.: Heiv. Chim. Acts 32, 215 (1949).
(303) SQUIBB AND SONS, E. R.: British patent 695,391 (1953); Chem. Abstracts 48, 9848 (1954).
(304) STECK, E. A., AND BOEHME, W.: J. Am. Chem. Soc. 74,4511 (1952).
(305) SuETA, H., AND MAEDA, A.: Japanese patent 1265 (1953);
Chem. Abstracts 48, 1864 (1954).
(306) SUGASAWA, S., AND SHIGEHARA, H.: Pharm. Soc. Japan 63, 98 (1943); Chem. Abstracts 44, 7310 (1950).
(307) SUGASAWA, S., AND SHIGEHARA, H.: Ber. 74, 459 (1941). (308) SUGINO, K., AND HAYASHI, S. S.: J. Chem. Soc. Japan 65, 458 (1944).
(309) SUGINO, K., ODo, K., AND SHIRAI, K.: J. Chem. Soc. Japan, Pure Chem. Sect. 71, 396 (1950).
(310) SUGINO, K., AND SEKINE, T.: J. Electrochem. Soc. Japan 104, 497 (1957);; Chem. Abstracts 51, 15307 (1957).
(311) SUGINO, K., AND SEKINE, T.: Japanese patent 4962 (1959); Chem. Abstracts 54, 5298 (1960).
(312) SUGINO, K., AND SHIRAI, K.: J. Chem. Soc. Japan, Pure Chem. Sect. 70, 111 (1949).
SUGINO, K., SHIRAI, K., SEKINE, T., AND ODo, K.: J. Electrochem. Soc. 104, 667 (1957).
(314) Sugino, K., AND YAMASHITA, M.: Japanese patent 172,275 (1946); Chem. Abstracts 43, 6096 (1949).
(315) SUGINO, K., AND YAMASHITA, M.: Japanese patent 175,935 (1948); Chem. Abstracts 44, 9836 (1950).
(316) SUGINO, K., AND YAMASHITA, M.: J. Chem. Soc. Japan, Pure Chem. Sect. 70, 71(1949).
(317) SUGINO, K., AND YAMASHITA, M.: Japanese patent 3926 (1955); Chem. Abstracts 51, 12712 (1957).
(318) SUPNIEWSKI, J., AND DANYSZ, A.: Polska Akad. Umiejetnosci, Rozprawy Wydzialu Lekarsk Ser. 1, 11, No. 9 (1950); Chem. Abstracts 45, 1593 (1951).
(319) SuzuKi, S., SHIMIzu, K., MORITA, M., AND UNO, K.: Japanese patent 2473 (1958); Chem. Abstracts 53, 5151 (1959).
(320) SWANN, S., JR.: Ind. Eng. Chem. 29,1339 (1937).
(321) SWANN, S., JR.: Trans. Electrochem. Soc. 84, 165 (1943). (322) SWANN, S., JR.: Bibliography of Electro-organic Chemistry,University of Illinois Bull. 45, No. 69, Circular Series
No. 50 (1948).
(297)
(313)
(323) SWANN, S., JR.: in Technique of Organic Chemistry, edited by A. Weissberger, Vol. II, 2nd edition. Interscience Publishers, Inc., New York (1956).
(324) SWANN, S., JR.: Bull. Central Electrochem. Res. Inst. 2, No. 1, 6 (1955).
(325) SWANN, S., JR., AMBROSE, P. E., DALE, R. C., RowE, R. C., WARD, A. W., KERFMAN, H. D., AND AXELROD, S.: Trans. Electrochem. Soc. 85, 231 (1944).
(326) SWANN, S., JR., BENOLIEL, R. W., LYONS, L. B., AND
PART, W. H.: Trans. Electrochem. Soc. 79, 83 (1940). (327) SWANN, S., JR., BRIGGS, S. W., NEKULTIN, V. C., AND JEROME, A. J.: Trans. Electrochem. Soc. 80, 163 (1941). (328) SWANN, S., JR., CHEN, C. Y., AND KERFMAN, H. D.: J. Electrochem. Soc. 99, 460 (1952).
(329) SWANN, S., JR., AND KERFMAN, H. D.: Trans. Electrochem. Soc. 92, 427 (1947).
(330) SWANN, S., JR., ONSTOTT, E. I., AND BAASTAD, F. H.: J. Electrochem. Soc. 102, 113 (1955).
(331) SWANN, S., JR., WANDERER, K. H., SCHAFFER, H. J., AND STREAKER, W. A.: J. Electrochem. Soc. 96, 353 (1949).
(332) SZMDRAGD, S., AND BRINER, E.: Hely. Chim. Acta 32, 553 (1949).
(333) TAFEL, J.: Z. physik. Chem. 34, 187 (1900). (334) TAFEL, J.: Ber. 33, 2209 (1900).
(335) TAFEL, J.: Z. physik. Chem. 50, 641(1905). (336) TAFEL, J.: Ber. 39, 3626 (1906).
(337) TAFEL, J.: Ber. 45, 3321 (1912).
(338) TAFEL, J., AND EMMERT, B.: Z. Elektrochem. 17, 569 (1911).
(339) TAFEL, J., AND WEINSCHENK, A.: Ber. 33, 3370 (1900). (340) TAHAGI, S., SuzuKI, T., AND DATE, J.: J. Pharm. Soc. Japan 71, 126 (1951); Chem. Abstracts 45, 6091 (1951). (341) TAHAGI, S., SuzuKI, T., AND INAEDA, K.: J. Pharm. Soc. JAPAN 69, 358 (1949); Chem. Abstracts 44, 1832 (1950). (342) TAMMANN, G.: Z. anorg. Chem. 126,176 (1923). (343) TANFORD, C., AND WAWZONEK, S.: Ann. Rev. Phys. Chem. 3, 247 (1952).
(344) TER-MINASYAN, L. E.: Zhur. Fiz. Khim. 27, 719 (1953). (345) TER-MINASYAN, L. E.: Izvest. Akad. Nauk Armyan. S.S.R., Ser. Khim. Nauk 10, 173 (1957); Chem. Abstracts 52, 2613 (1958).
(346) TER-MINASYAN, L. E.: Doklady Akad. Nauk Armyan.
S.S.R. 25, 243 (1957); Chem. Abstracts 52, 9814 (1958). (347) TER-MINASYAN, L. E.: Izvest. Akad. Nauk Armyan.S.S.R., Ser. Khim. Nauk 11, 75 (1958); Chem. Abstracts 52, 18022 (1958).
(348) TER-MINASYAN, L. E.: Izvest. Akad. Nauk Armyan. S.S.R., Ser. Khim. Nauk 11, 221 (1958); Chem. Abstracts 53, 5916 (1959).
(349) ToMILOv, A. P., AND KAABAK, L. V.: Zhur. Priklad. Khim. 32, 2600 (1959); Chem. Abstracts 54, 7374 (1960).
(350) UEDA, T., NAGATA, I., AND ITO, S.: Pharm. Bull. 1, 322 (1953).
(351) UDUPA, H. U. K., AND DEY, B. B.: Proc. 6th Meeting Intern. Comm. Electrochem. Thermodynam. and Kinet. 1955, 465; Chem. Abstracts 50, 6221 (1956).
(352) UDUPA, H. U. K., AND DEY, B. B.: Indian patent 60,864 (1959); Chem. Abstracts 54, 7381 (1960).
(353) URABE, N., AND YASUKACHI, K.: J. Electrochem. Soc. Japan 22, 525 (1954); Chem. Abstracts 49, 9398 (1955).
(354) VASIL'EV, S. V., AND VOVCHENKO, G. D.: Vestnik Moskov. Univ. 5, No. 3, Ser. Fiz: Mat. i Estest. Nauk, No. 2, 73 (1950); Chem. Abstracts 45, 6594 (1951).
(355) VERTYULINA, L. N., AND MALYUGINA, N. I.: Zhur. Obshchel Khim. 28, 304 (1958).
(356) VINOGRADOVA, A. I., AND ARKHANGELSKAYA, V. N.: Zhur. Obshchel Khim. 16, 301 (1946).
(357) VODZINSKII, Y. V., AND KORSHUNOV, I. A.: Uchenye Zapiski Gor'kovsk. Gosudarst. Univ. im. N. I. Lobachev skogo, Ser. Khim. 1958, No. 32, 25; Chem. Abstracts 54, 17113 (1960).
(358) VOITKEVICH, S. A.: Zhur. Fiz. Khim. 26, 869 (1952). (359) WARTHA, V.: Chem. Ztg. 8, 431 (1884). (360) WAWZONEK, S.: Anal. Chem. 32, 144R (1961).
(361) WEBER, J. E., AND MEISTER, A. E.: J. Chem. Educ. 27,571 (1950).
(362) WENKER, H.: J. Am. Chem. Soc. 57, 772 (1935).
(363) WEYGAND, C., GABLER, A., AND BIRCAN, N.: J. prakt. Chem. 158, 266 (1941).
(364) WIBAUT, J. P., AND BOER, H.: Rec. tray. chim. 68, 72 (1949).
(365) WIELAND, H., AND JENNER, R. G.: Ann. 545, 86 (1940). (366) WIELAND, H., AND LIEBIG, R.: Ann. 555, 146 (1944). (367) WIELAND, H., AND MULLER, 0.: Ann. 545, 59 (1940). (368) WILSON, C. L.: Trans. Electrochem. Soc. 75, 353 (1939).
(369) WILSON, C. L.: Trans. Electrochem. Soc. 92, 369 (1947). (370) WILSON, C. L., AND UDUPA, H. V.: J. Electrochem. Soc. 99, 289 (1952).
(371) WILSON, C. L., AND WILSON, K. L.: J. Chem. Soc. 1941, 874.
(372) WILSON, C. L., AND WILSON, K. L.: Trans. Electrochem.
Soc. 80,139 (1941). WILSON, C. L., AND WILSON, K. L.: Trans. Electrochem. Soc. 80, 151 (1941).
(374) WILSON, C. L., AND WILSON, K. L.: Trans. Electrochem. Soc. 84, 153 (1943).
(375) WITTIG, G., AND FARTMANN, B.: Ann. 554, 229 (1943). (376) WOLFROM, M. L., BINKLEY, W. W., SPENCER, C. C., AND LEW, B. W.: J. Am. Chem. Soc. 73, 3357 (1951).
(377) WOLFROM, M. L., KONIGSBERG, M., MOODY, F. B., AND GOEPP, R. M.: J. Am. Chem. Soc. 68, 122 (1946).
(378) WOLFROM, M. L., LEW, B. W., HALES, R. A., AND GOEPP, R. M.: J. Am. Chem. Soc. 68, 2342 (1946).
(379) WOLFROM, M. L., MOODY, F. B., KONIGSBERG, M., AND GOEPP, R. M.: J. Am. Chem. Soc. 68, 578 (1946).
(380) WOLK, I. L.: U.S. patent 2,419,515 (1947); Chem. Abstracts 42, 589 (1948).
(381) WOODRUFF, E. H.: J. Am. Chem. Soc. 64, 2859 (1942). (382) YAMAMOTO, S., NOBUKUNI, T., AND MATSUDA, M.: Japanese patent 1867 (1958); Chem. Abstracts 53, 5151 (1959).
(383) YAMASHITA, M.: J. Electrochem. Soc. Japan 21, 376 (1953); Chem. Abstracts 48, 13484 (1954).
(384) YAMAZAKI, T., AND NAGATA, M.: Yakugaku Zasshi 79, 1222 (1959); Chem. Abstracts 54, 4596 (1960).
(385) ZERBES, G.: Z. Elektrochem. 18, 619 (1912).
(386) ZWICKER, B. M. G., AND ROBINSON, R. J.: J. Am. Chem. Soc. 64, 790 (1942).
Chemistry is our Covalent Bond
(Hive Addict)
06-16-03 21:48
No 440450
Very informative.. Lugh, have you ever gotten an electrochemical reduction to work??? I have yet to meet anyone who has...
Infinite Radiant Light - THKRA
(Hive Bee)
06-17-03 12:16
No 440586
I read somewhere that the reduction of amides to amines happens doing a catalytic hydrogenation , in a non aqueous solvent using Raney nickle as the catalyst under nl hydrogenation.....java
Sorry no references!
We're all in this world together,
http://www.chiapaslink.ukgateway.net/
(Hive Addict)
06-17-03 12:41
No 440592
Yes amines can be made from amides by catalytic hydrogenation, but the pressures required are way beyond the capability of 99.99% of all bees. We're talking about a couple of hundred bar at least.
Freaky
(Hive Bee)
06-19-03 11:19
No 441077
I asked someone about this problem with hydrogenation of amides to amines , this is what I was told,
'"First the methyl amide on hydrogenation will give ethyl amine and ethyl amide will give propyl amine. The temperatures would be around 60 degrees centigrade or thereabouts. I you are restricting H2 pressures to 60 psi, go by exotherm as guideline for fixing temperature or by H2 consumption. Similarly catalyst concentration will play a role in determining temperature of the reaction."
I will try to get some references for this statement and how to limit the process to methylamine............java
We're all in this world together,
http://www.chiapaslink.ukgateway.net/
(Chief Bee)
06-28-03 11:41
No 443146
(Rated as: good read)
According to Post 436887 (Baba_McKensey: "amides to amines", Serious Chemistry) the following article is required reading when it comes to electrolytic reduction of amides to amines:
Bull. Chem. Soc. Japan 11, 41 (1936) (../rhodium/pdf /amide2amine.
(Moderator)
06-28-03 13:18
No 443153
(Rated as: good read)
The actual preparation of the cathode is in Bulletin of the Chemical Society of Japan 7 156 (1932):
The cathode of zinc amalgam was prepared in the following way:
Into a cylindrical cell having a diameter of 4.5 cm. mercury was introduced to the depth of about 1 cm., and after a conducting wire sealed up in a small glass tube was inserted into mercury a solution of zinc sulphate was poured into the cell as the catholyte. The cell was then placed in a glass cylinder which contained sulphuric acid of 6 normal as the anolyte. Now taking a lead plate as the anode a current of 2 amperes was passed between this and the mercury, which served as the cathode. The electrolysis was continued till the mercury completely solidified as zinc amalgam.
Chemistry is our Covalent Bond
(Hive Addict)
07-13-03 04:49
No 446794
(Rated as: good read)
#1 method besides LAH is Borane-THF. Stir it up in borane-THF and then quench with MeOH and evaporate from MeOH a few times. Trimethyl Borate azeotropes with MeOH at ~50°C. Unfortunatly, BH3 is moisture sensitive and a fire hazard.
#2- Lithium Borohydride. Stir it up in alcohol, or ethers. LiBH4 is expensive, but can be made in situ from NaBH4 + LiCl (excess). Actually, you can make Borane in situ from NaBH4 plus any lewis or protic acid. TMS-Cl, BF3, etc. Or you can form Nickel or Zinc borohydrides by addition of the metal salt. (ZnCl2, NiCl4, etc.)
I don't think that Catalytic hydrogenation is not a realistic method for reducing amides. Amides are very easy functional groups to make and very stable, hence nature's extensive use of them. But they are hard to reduce/hydrolyse/dehydrate etc.
(Hive Bee)
07-14-03 07:46
No 447088
To follow up on the catalytic hydrogenation conversation using Pd/charcoal to reduce amides to amines....
.".... sorry for the delay, I had been out of station for office work and so could not go to library to check these old references. Now I have gone thru them, and feel that you try using Platinum or better Palladium catalyst on charcoal support, the details will be available on Johnson-Matthey site or any other noble metal catalyst manufacturers. Normal procedure will be to use suitable solvent like methanol/ethanol/isoprpyl alcohol and about 0.5 gms of catalyst ( loading 5%) per gm.mole of starting amide and follow temperature/pressure profile at which you get exotherm. Limit pressures to 3-4 kg/cm2g as you desire and increase temperature to max. 100 degrees centigrade."
I was not given any references to quote but only the comments from a hands on researcher ......I hope this sheds some light, on the catalytic hydrogenation of amides >>>amines at low pressure<60 psi. with temp max 100 C using Pd/charcoal although if one starts with a methyl amide the product will be an ethylamine, which can still be used to aminate the favorite ketone...java
We're all in this world together,
http://www.chiapaslink.ukgateway.net/
(Hive Addict)
07-16-03 03:48
No 447756
(Rated as: good read)
You cannot reduce amides with Palladium on Carbon under any normal conditions. Maybe 0.00001% would end up reduced, but that's it. I have taken amides and polyamides from 40psi-500psi with copious amounts of fresh 10% Pd/C and never had any reduction. With PtO2 in acid I would beliveve that that could that happen though.
Besides you need to have an acidic enviroment to dehydate the hemiaminal formed. -C(=O)N- -> -CH(OH)N- -(-H2O)-> -CH=N+- -> -CH2-NH-. When you reduce with a borohydride or LAH, you have borane or alane present which are lewis acids.
(Hive Bee)
11-04-03 09:54
No 468717
(Rated as: excellent)
Lego's idea is as follows:
1) Convert the amide to a thioamide with any known reagent. Lego would suggest P4S10 or Lawesson's reagent (p-MeOC6H4)2P2S4). The first is cheap and yields are OK, the second is a bit more expensive but yields are usually better.
Just a few interesting examples:
Organic Letters, 1999, 1(5), 697-700
DOI:10.1021/ol990629a
Microwave-Accelerated Solvent-Free Synthesis of Thioketones, Thiolactones, Thioamides, Thionoesters, and Thioflavonoids
An expeditious, solvent-free, and high yield conversion of ketones, flavones, isoflavones, lactones, amides, and esters to the corresponding thio analogues is described utilizing Lawesson's reagent in a process that circumvents the use of dry solvents and excess of the reagent.
Synthetic Communications, 1990, 20(19), 3085-3095
In situ reagents for thionation of amides, peptides and lactams
An agent, prepd. from 1:1 P2S5 and Na2CO3 in situ, is used for thionation of amides, amino acids, peptides, and lactams. Yields range from 28 to 96%.
The latest article on this topic (especially check the reference mentioned in the beginning):
J. Org. Chem., 2003, 68(14), 5792-5794
DOI:10.1021/jo0344485
Mild Method for the Conversion of Amides to Thioamides
Aqueous ammonium sulfide was found to be an ideal substitute for hydrogen sulfide for the thiolysis of activated amides. High yields of the corresponding thioamides were obtained for a broad range of substrates, using two different procedures that are both operationally simple and inexpensive, as well as amenable to large-scale preparation. Preliminary results indicate that aqueous ammonium sulfide may also replace hydrogen sulfide in the synthesis of thionoesters from amides.
There are so many more articles concerning this topic, but this is enough to give you an idea of how this versatile this reaction is.
2) Reduction of the thioamide to an amine.
As Al/Hg is a common reducing agent in clandestine chemistry here is a reference:
Ber., 1921, 54B, 1080-1081
Thioamides. II. Reduction of thioamides to primary amides.
By the use of Al-Hg in a neutral medium thioamides can be reduced to the corresponding primary amines with good yields. Thus, 0.1 mol. PhCSNH2 in 4 parts 99% alc. and 13 g. Al-Hg slowly treated under a reflux with turbining with 13 cc. H2O, heated 3 hrs. at 60-70°, made alk. with NaOH, distd. with steam, acidified with HCl, concd., decompd. with concd. KOH, taken up with Et2O and dried with K2CO3 give PhCH2NH2, b. 184.5°. MeCSNH2 (1 g.) in 10 cc. of 90% alc. similarly reduced with 1 g. Al-Hg and 1 cc. H2O gives EtNH2.
Or take a look in Chemical Reviews, 1961, 61, 45-86. Electrolysis, catalytic hydrogenation, metal/acid-combinations can also be used to reduce thioamides.
If there is any further interest on this kind of reaction Lego will post explicit experimental details and search this fucking reference where a thioamide was reduced in 90% yield with Zn/HOAc.
Ooops, while looking for the exact references Lego found an article with a similiar, more sophisticated reaction.
Tetrahedron Letters, 1980, 21(42), 4061-4064
A convenient method for the selective reduction of amides to amines
Amides were selectively reduced to the corresponding amines in high yield via their thioamides and (alkylthio)methyleniminium salts. E.g., PhCH2CONMe2 on treatment with (p-MeOC6H4)2P2S4 (PhMe, under Ar, 100°, 4 h) gave 90% PhCH2CSNMe2. The iminium salt obtained by treating the latter with Et3O+ BF4- (CH2Cl2, under Ar, 0°, 5 min, then room temp., 45 min), was reduced quant. with NaBH4 (MeOH, 0°, 5 min, then room temp., 2 h) to Ph(CH2)2NMe2. This procedure is compatible with isolated and conjugated double bonds, esters, nitro groups, and sulfonamides.
The candle that burns twice as bright burns half as long
(Heavyweight Chempion(eer))
11-05-03 04:33
No 468904
Raney nickel is known to easily rip sulfur from organic compounds to form NiS irreversily. Perhaps urushibara nickel, in some form, have the same properties. With Raney nickel it is just a matter of allowing the thio compound to react with a slight excess nickel at a slightly elevated temperature and in a suitable solvent. Voila, a desulfurised compound.
(Hive Bee)
11-13-03 09:59
No 470674
(Rated as: excellent)
Here is the reference for the reduction of a secondary thioamide with Zn/HCl in EtOH (yield: 91%).
http://www.unibas.ch/mdpi/ecsoc-3/a0020/
More on this topic will follow when Lego's alter ego will not bee busy during the opening times of the library.
N-METHYLTYRAMINE AND N,O-DIMETHYLTYRAMINE SYNTHESIS VIA INTERMEDIATE 4-METHOXYPHENYL-N-METHYLTHIOACETAMIDE
Vladimir N. Bulavka*, Alexander N. Shchavlinskii and Oleg N. Tolkachev
All-Russian Institute of Medicinal and Aromatic Plants (VILAR);
Grina str., 7, Moscow, 113628, Russian Federation.
*Present address: Scientific-Research Phototechnical Institute on Slavich Company (NIFTI-Slavich);
Mendeleev sq. 2, Pereslavl-Zalesskii, Yaroslavl region, 152140, Russian Federation.
E-mail: nifti@slavich.botik.ru
Received: 30 July 1999 / Uploaded: 13 August 1999
Keywords: galanthamine synthesis half-products, N-methyltyramine, N-methyltyramine ethers, N,O-dimethyltyramine, arylthioacetamide, Willgerodt-Kindler reaction
N-Methyltyramine and its alkyl or benzyl ethers are common intermediate products in the syntheses of narwedine, and galanthamine alkaloids [1-8]. They are usually produced from the corresponding arylacetamides by lithium aluminium hydride reduction. In order to avoid application of inflammable lithium aluminium hydride bulgarian scientists have transformed arylacetamide to arylthioacetamide with phosphorus (V) sulfide with following reduction of arylthioacetamide with complex sodium borohydride - nickel or cobalt chloride agents [7, 8].
Here we present a new short way route to N-methyltyramine and its ethers via the corresponding aryl-N-methylthioacetamides, shown at the scheme 1 at the example of N-methyltyramine and N,O-dimethyltyramine synthesis.
Scheme 1.
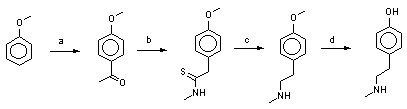
--> a: 92%
--> b: 57-58%
--> c: 91%
--> d: 98%
total 46-47% total 48-49%
a) H3CCOBr, AlCl3, ClCH2CH2Cl.
b) H3CNH2.HCl, S8 , H3CCO2Na, (H3C)2NCHO, 115oC, 3h.
c) Zn (dust) and 36% HCl(aq.) in ethanol
d) 36% HCl(aq.), refluxion
Anisole was smoothly acetylated in a classical conditions [9] with acetyl bromide and aluminium chloride in dichloroethane to produce 4-methoxyacetophenone in 92% yield.
Phenylacetic acid thioamides are usually synthesized by the Willgerodt-Kindler reaction from the corresponding substituted acetophenones, amines, and sulfur. We have studied several alternative routes to the 4-methoxyphenyl-N-methylthioacetamide synthesis shown at the scheme 2.
Scheme 2.
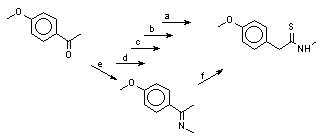
a) H3CNH2, S8, 170-180oC, 3h (yield 10,4%, and 21,9% by-product amide).
b) H3CNH2.HCl, S8 , H3CCO2Na, (H3C)2NCHO, 115oC, 3h (yield 58,4%).
c) H3CNH2.HCl, S8, N(C2H5)3, 4-H3CC6H4SO3H.H2O, (H3C)2NCHO, 75oC, 72h (yield 57%)
d) H3CNHCHO, S8, 190oC, 20h (yield 35%).
e) H3CNH2 .HCl, Na2CO3, CaO, N(C2H5)3, -7oC (isolated mixture imine : ketone 77:23 (1H NMR) formed, used for the next stage without separation).
f) S8, N(C2H5)3, (H3C)2NCHO, 70oC (total yield on two stages 21,5%).
The best results were produced according to routes b and c.
The key thioamide was isolated chromatographically on alumina or silica gel.
In classical conditions of the Willgerodt-Kindler reaction in the sealed tube [10] the desired thioamide was obtained in only 10.4% yield, while the corresponding by-product amide was also isolated in 21.9% yield. In order to avoid the application of high pressure, two-stage synthesis was carried out via intermediate 4-methoxyacetophenone methylimine [11] by method [12, 13] in 21.5% total yield.
Earlier proposed method of carrying out the Willgerodt-Kindler reaction with volatile lower secondary amines, e. g. dimethylamine, using its hydrochloride and sodium acetate in dimethyl formamide at 100-110oC and atmospheric pressure was developed [14].
We have used a similar method for lower primary amine. Thus methylamine hydrochloride, 4-methoxyacetophenone, sulfur, and a base in dimethyl formamide at 60-110oC and atmospheric pressure gave the desired thioamide in 57-58% yield.
An other route of 4-methoxyphenyl-N-methylthioacetamide synthesis on heating the mixture of 4-methoxyacetophenone, N-methylformamide and sulfur at 170-180oC at atmospheric pressure according to the method [15] 35% yield was achieved.
4-Methoxyphenyl-N-methylthioacetamide is a new compound, forming colorless crystals, m. p. 89-91oC (from ethanol or from diethyl ether) [16].
We have elaborated for the thioamide obtained a new simple and efficient method of reduction with zinc dust and hydrochloric acid in ethanolic solution. N,O-Dimethyltyramine was obtained in 91% yield, which was converted to its hydrochloride, m.p. 179-181oC (from ethanol), (lit. m.p. 181-182oC [17]).
O-Demethylation of N,O-dimethyltyramine to N-methyltyramine was carried out according to earlier described method for N-acetylated compound [18] by refluxing with concentrated hydrochloric acid in the oil bath heated to 170-175oC. The yield was 98%, m. p. 146-149oC (from ethanol with a
(Hive Bee)
11-13-03 11:25
No 470682
Great find Lego!
Someone has to try it on a more interesting substrate!
1,4-dimethoxybenzene is a good choice.
(Hive Bee)
01-08-04 18:50
No 481329
See
Post 311068 (PolytheneSam: "Electrochemical reduction", Tryptamine Chemistry)
N'ętre que de la drogue
(Stranger)
02-21-04 14:40
No 490220
In column 4 lines 17+ of
Patent US5074974
it says
an oxidation synthesis can be conducted in an electrochemical cell divided by an anion-exchange membrane. In this instance, the anolyte will be the working electrolyte, and anions will selectively flow through the membrane from the catholyte (counter electrolyte) into the anolyte. Or, alternatively, a reduction synthesis can be conducted in a cation-exchange membrane-divided cell. In this example, the catholyte will be the working electrolyte, and cations will be be selectively exchanged from the anolyte (counter electrolyte) through the membrane to the catholyte (working electrolyte).
US 5074974 has the same inventor listed as the patent here:
Post 312706 (PolytheneSam: "US 4,695,352", Tryptamine Chemistry)
(Stranger)
02-21-04 14:57
No 490226
Note the use of titanium here:
Patent US742797
Patent US4695352
(Hive Bee)
06-28-04 14:24
No 516067
I have been looking for a specific method to remove the amide formed on Phenylalalnie via acylation that is being used as a protecting group while the COOH is being reduced to CH3. I've considered the -OBr method but it's limited to aliphatics and the catalytic reduction seems not doable with my Parr Hydrogenator that is limited to 60 p.s.i.. in reducing the amides without making the amide a better leaving group. By doing it this way it leave a methyl amine hence the finish product is a meth-amphetamine as opposed to just amphetamine
I've read the methods suggested by Lego and others in converting the amide to easier leaving groups, thioamides, that can be removed easily but need ....
The Preparation and Chemical Properties of Thionamides.
Richard N. Hurd, George DeLaMater;
Chem. Rev.61(1); 45-86,1961
this method might work and the removal by catalytic hydrogenation appeals to me....java
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/
(Hive Bee)
06-30-04 10:37
No 516568
As I searched some of the old threads where I found a posting by Post 348699 (Cyrax: "Li/Na Borohydride and Trimethylchlorosilane", Serious Chemistry) where he quotes a text from Rhodium....
Lithium/Sodium Borohydride and Trimethylchlorosilane - An Unusually Strong and Versatile Reducing Agent
Angew. Chem. Int. Ed. Engl. Vol 28, No. 2, 218-220 (1989) (../rhodium /nabh4-
where they use procedure A on example 7, where the amide is reduced to an amine , hence the procedure will work for the reduction of the acylated amine in phenylalanine once the COOH is reduced ,....java
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/
(Moderator)
07-03-04 17:36
No 517284
I've read the methods suggested by Lego and others in converting the amide to easier leaving groups, thioamides, that can be removed easily but need ....
The Preparation and Chemical Properties of Thionamides.
Richard N. Hurd, George DeLaMater;
Chem. Rev.61(1); 45-86,1961
Chem. Rev.61(1); 45-86,1961
![]()
Chemistry is our Covalent Bond
(Chief Bee)
11-03-04 17:04
No 539539
(Rated as: good read)
Zinc Borohydride: Reduction of Amides to Amines
S. Narasimhan, S. Madhavan, R. Balakumar & S. Swarnalakshmi
Synth. Commun. 27(3), 391–394 (1997) (../rhodium /amide2
Abstract
Zinc borohydride reduces secondary amides to the corresponding N-ethyl amines in excellent yields.
The reduction requires only stoichiometric quantities of hydride and does not require the addition of any Lewis acid.
The amides are isolated by simple hydrolysis of the reaction mixture.
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
11-09-04 12:32
No 540691
(Rated as: excellent)
An efficient sequence for the preparation of small secondary amine hydrochloride salts for focused library generation without need for distillation or chromatographic purification
Gene M. Dubowchik, Jodi A. Michne and Dmitry Zuev
Bioorg. Med. Chem. Lett., 2004, 14 , 3147-3149
DOI:10.1016/j.bmcl.2004.04.014
Abstract: Collections of small secondary amines for compound library generation can be efficiently prepared by amide reduction using BH3–THF or Red-Al followed by brief methanolysis, trapping with di-tert-butyl dicarbonate, and deprotection with 4M HCl in dioxane. The sequence requires no chromatography or distillation and provides multi-gram quantities of pure HCl salts in a short time.
Reagents and conditions:
(a) R2NH2, Et3N, CH2Cl2, 0°C to rt
(b) (i) BH3–THF (3 equiv), reflux, 14 h;(ii) MeOH, reflux, 2 h; (iii) Boc2O (1.4 equiv), CH2Cl2, rt, 14 h
(c) 4M HCl/dioxane (1.2 equiv), CH2Cl2, rt, 14 h.
Representative synthetic procedure for 4c:
A stirred solution of 3,3,3-trifluoroacetic acid N-hydroxysuccinimide ester (12.98 g, 57.65 mmol) in CH2Cl2 (80 mL) at 0 °C was treated with cyclopropylmethylamine (5.0 mL, 1 equiv). The mixture was stirred at rt for 14 h and then concentrated in vacuo. The residue was partitioned between ethyl acetate and water. The organic phase was washed with water, brine, dried over MgSO4, and evaporated to give the crude amide. This was dried under high vacuum for several hours and then, under nitrogen at 0 °C, it was carefully treated with 1M BH3 in THF (173 mL, 3 equiv). The mixture was heated at reflux for 14 h and then re-cooled to 0 C. MeOH (50 mL) was added carefully to avoid excess foaming, and the mixture was heated at reflux for 5 h. Upon re-cooling to 0 C, a solution of Boc2O (17.62 g, 1.4 equiv) in CH2Cl2 (25 mL) was added. The resulting mixture was stirred at rt overnight and then concentrated in vacuo. The residue was partitioned between ethyl acetate and water.
The organic was washed with water, brine, dried over MgSO4, and evaporated to give the crude Boc-protected amine. This was dissolved in CH2Cl2 (25 mL) and treated with 4M HCl in dioxane (17 mL, 1.2 equiv), carefully to avoid uncontrolled bubbling. The mixture was stirred at RT overnight and then evaporated. The resulting white solid was triturated with ether and the product was collected by filtration, washed with ether, and dried in vacuo (10.10 g, 86%).
The tendency is to push it as far as you can