(Hive Addict)
08-03-03 09:40
No 451451
(Rated as: excellent)
The following article has been requested by Chimimanie and retrieved by Hyberlab Bee azole: K Bodendorf, A Walk. Darstellung und Reduktion von Indolyl-3-aminomethylketonen. Arch Pharm 294(8) (1961) 484-487 (../rhodium/djvu /bodendorf.d
SYNTHESIS AND REDUCTION OF INDOLYL-3-AMINOMETHYLKETONES
Experimental
Indolyl-3-tert.butyl-carbinol
A solution of 9.6 g (0.4 mol) magnesium and 40 g (0.4 mol) t-BuCl in 200 mL THF was added to a solution of 14.5 g (0.1 mol) indolyl-3-aldehyde in 50 mL THF (good stirring necessary). When the addition was complete, the reaction mixture was refluxed for another hour and the solvent removed via vacuum distillation. The residue was treated with NH3/NH4Cl solution and extracted with ether. A brownish green substance was obtained after removal of the ether, but further purification with petroleum ether was possible. The substance gave colourless needles from water, mp 119-120°C. Yield: 13.5 g = 66.5%.
The same compound can also be obtained by reaction indolyl-magnesium bromide with trimethylacetaldehyde, but it suffers from low yields (only 25%).
Indolyl-3-bromomethylketone
15.9 g (0.1 mol) indolyl-methylketone was suspended in 50 mL methanol. Under stirring and cooling, 16 g (0.1 mol) bromine was added dropwise; doing so, the ketone dissolved completely. When the addition was complete, the reaction mixture was refluxed for 1-2 hours on the waterbad. The precipitation of brick-red crystals from the dark red solution was noted; the crystals were removed via suction filtration, washed with ether and recrystallized from methanol. The mp and mixed mp with authentic sample was 230°. Yield: 17.8 g = 74.8%
Indolyl-3-dimethylaminomethylketone (II)
11.9 g (0.05 mole) indolyl-bromomethylketone was dissolved in 60 mL hot IPA and refluxed on the waterbath for 1 hour after addition of 17.8 g (0.15 mol) 38% aqueous dimethylamine solution. When the reaction was halted, the volume was reduced to about half and colourless crystals started to form. They were isolated via suction filtration and recrystallized from ethanol; mp: 208-209°C. Yield: 8.5 g = 84.2%. mp HCl salt: 229°C. mp picrate salt: 181-183°C.
Indolyl-3-diethylaminomethylketone (III)
In analogy to the synthesis of II, we obtained said compound from 11.9 g indolyl-bromomethylketone and 11.0 g diethylamine in IPA. After recrystallization from ethanol, the mp was 136-137°C. Yield: 9.8 g = 85.3%. mp of the HCl salt: 211°C.
Indolyl-3-piperidinomethyl-ketone (VI)
In analogy to II, we obtained said compound from 11.9 g indolyl-bromomethylketone and 12.8 g piperidine in IPA. The mp was 169-170°C (after recrystallization from 50% alcohol). mp of the HCl salt: 214°C. Yield: 9.1 g (75.2%).
Indolyl-3-morpholinomethylketone (V)
In analogy to II, we prepared said compound from 11.9 g indolyl-bromomethylketone and 13.0 g morpholine in IPA. Its mp was 167°C (HCl salt: 218°C) after recrystallization from ethanol.
Indolyl-3-methylaminomethylketone (VI)
In analogy to II, said compound was prepared from 11.9 g indolyl-bromomethylketone and 47 g 33% methylamine solution in IPA. The mp was 197-198°C (recrystallized from ethanol; mp HCl salt: 235-237°C). Yield: 5.5 g = 58.5%.
Indolyl-3-aminomethylketone hydrochloride (VII)
11.9 g indolyl-bromomethylketone was dissolved in a small amount of acetone and added to 100 mL chloroform. A solution of 7.0 g (0.05 mol) hexamine in 100 mL was added, and the mixture allowed to react overnight. The white and voluminous precipitate was isolated via suction filtration and dried. mp of the adduct: 230-232°C. Yield: 18.9 g = 100%.
A cold mixture of 15 mL 38% HCl and 120 mL ethanol was poured over the adduct, and the product set aside at room temperature for three days (with occasional shaking). The reaction mixture's volume was reduced in vacuo and the residue recrystallized from water.
Residual bromide ions were trapped by dissolving the obtained substance in hot water and strong stirring (1 h) after addition of AgCl. The solvent was damped of after filtration of the silver halogenide, after which colourless needles of the HCl salt precipitated; mp: 268-270°C. Yield: 8.5 g = 73.9%.
1-Indolyl-3-(2)-dimethylaminoethane (VII)
A suspension of 2.3 g (0.06 mol) LAH in 50 mL THF was added to a solution of 4.0 g (0.02 mol) indolyl-dimethylaminomethylketone in 30 mL THF. The mixture was stirred for 30 minutes at room temperature, followed by 2 hours on a boiling waterbath. The THF was removed via distillation and water added to the complex. By doing so, a smudgy mixture of base and aluminium hydroxide was obtained. It was extracted with acetone after decanting the water; when the acetone was dried over potassium carbonate, it was evaporated, hereby giving a brownish oil. The oil crystallized after purification with ether-petroleum ether. Recrystallization from ethyl acetate-petroleum ether gave the compound, mp 48-49°C. Yield: 2.8 g = 73.7%. mp of the picrate salt: 170°C.
1-Indolyl-3-(2)-methylaminoethane (IX)
A suspension of 3.8 g (0.02 mol) indolyl-methylaminomethylketone in 30 mL THF was added to a suspension of 2.3 g (0.06 mol) LAH in 50 mL THF. The reaction was conducted as described for VIII. During the work-up, a yellow oil was obtained, which after purification with ether-petroleum ether crystallized; the recrystallized product (ethyl acetate-petroleum ether) had mp 89°C. Yield: 2.4 g = 68.6%. mp of the HCl salt: 175°C.
Dirty old man
(Hive Bee)
08-03-03 20:23
No 451545
Thank you Azole and GC!
Well, speaking about [1], we now have the yield in the bromination step: 75% and the yield in the swap step is improved (altough the method in the other article is inspired by this one), good!
Ok, lets see what the yields are from the alpha-chloro analogs with the other Arch Pham ref (Archiv der Pharmazie (Weinheim, Germany) (1978), 311(3), 248-55) (EDIT: Retrieved (Thanks again GC!
At least we have a good synth already from indole, acetonitrile (cheap as dirt
Nice
Good that LiAlH4 is not needed and can bee replaced by borohydride!
Good work, bees.
EDIT:
Thanks to GC-MS [4] we have now the yield for the Cl swap: 74%.
And now, let me...
...propose an Experimental:
So here is an example of a 3 steps synthesis of DET taken from the linked posts above:
indole + chloracetyl chloride + SnCl4 => 3-chloroacetyl-indole: [2]
To a stirring solution of indole (1.17 g, 10 mmol) in CH2Cl2 (20 mL) under argon at 0 °C was added SnCl4 (1.44 mL, 12 mmol) in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then chloro acetylchloride (10 mmol) was added in small portions to the suspension, followed by nitromethane (15 mL). The mixture was stirred for 8 h at room temperature. After being quenched with ice and water (30 mL), the mixture was filtered to remove inorganic precipitates, and the organic material was extracted with ethyl acetate (50 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to give the product as a crystalline solid.
Yield: 80%, mp: 235°C. [4]
3-chloroacetyl-indole => 3-(N,N-diethylaminoacetyl)-indole: [4]
7.7 g (0.04 mol) 3-chloroacetyl-indole was suspended in 300 mL abs toluene and refluxed for 2 hours with 8.8 g (0.12 mol) diethylamine. The solvent is evaporated, the reaction mixture cooled down and the precipitate collected and washed with water. The residual base was recrystallized from toluene. Formation of the HCl salt was achieved by means of gaseous HCl in methanol.
Yield: 74%, mp: 205-215°C.
3-(N,N-diethylaminoacetyl)-indole (keto-DET) => DET: [1]
The 3-(N,N-diethylaminoacetyl)-indole was refluxed with twice its weight of sodium borohydride in 1-propanol for 15 hours. The solvent was evaporated in vacuo and the residue was partitioned between water and chloroform. The chloroform extract was extracted with 3N-hydrochloric acid to separate the tryptamine from accompanying 3-unsubstituted indole. The acid extracts were alkalized with 10% sodium hydroxide and the liberated bases extracted into chloroform. The tryptamine was isolated either as the free base or picrate salt.
Yield: probably between 40-50% (49% for DMT but it seem the diethyl is a bit lower in yield), mp: 84-87 °C (freebase, Tihkal).
Alternatively, synthesis of DMT in 4 steps:
indole + SnCl4 and acetonitrile => 3-acetyl-indole: [2]
To a stirring solution of indole (1.17 g, 10 mmol) in CH2Cl2 (20 mL) under argon at 0 °C was added SnCl4 (1.44 mL, 12 mmol) was added in a single portion via syringe. After the ice bath was removed, the mixture was stirred at room temperature for 30 min, and then acetonitrile (100 mmol, 5.3 mL) was added in small portions to the suspension, followed by nitromethane (15 mL). The mixture was stirred for 4 h at room temperature. After being quenched with ice and water (30 mL), the mixture was filtered to remove inorganic precipitates, and the organic material was extracted with ethyl acetate (50 mL). The organic phase was dried over Na2SO4 and concentrated at reduced pressure to give the product as a crystalline solid.
Yield: 96%, mp: 185-191°C.
3-acetyl-indole => Indolyl-3-bromomethylketone: [3]
15.9 g (0.1 mol) indolyl-methylketone was suspended in 50 mL methanol. Under stirring and cooling, 16 g (0.1 mol) bromine was added dropwise; doing so, the ketone dissolved completely. When the addition was complete, the reaction mixture was refluxed for 1-2 hours on the waterbad. The precipitation of brick-red crystals from the dark red solution was noted; the crystals were removed via suction filtration, washed with ether and recrystallized from methanol. The mp and mixed mp with authentic sample was 230°.
Yield: 17.8 g = 74.8%, mp: 230°C.
Indolyl-3-bromomethylketone => Indolyl-3-dimethylaminomethylketone (keto-DMT) [3]
11.9 g (0.05 mole) indolyl-bromomethylketone was dissolved in 60 mL hot IPA and refluxed on the waterbath for 1 hour after addition of 17.8 g (0.15 mol) 38% aqueous dimethylamine solution. When the reaction was halted, the volume was reduced to about half and colourless crystals started to form. They were isolated via suction filtration and recrystallized from ethanol.
Yield: 8.5 g = 84.2%, mp: 208-209°C; mp HCl salt: 229°C; mp picrate salt: 181-183°C.
Indolyl-3-dimethylaminomethylketone (keto-DMT) => DMT
-with sodium borohydride: [1]
The 3-(N,N-dimethylaminoacetyl)-indole was refluxed with twice its weight of sodium borohydride in 1-propanol for 15 hours. The solvent was evaporated in vacuo and the residue was partitioned between water and chloroform. The chloroform extract was extracted with 3N-hydrochloric acid to separate the tryptamine from accompanying 3-unsubstituted indole. The acid extracts were alkalized with 10% sodium hydroxide and the liberated bases extracted into chloroform. The tryptamine was isolated either as the free base or picrate salt.
Yield: 49%, mp: 42-43°C.
-with lithium aluminium hydride: [3]
A suspension of 2.3 g (0.06 mol) LAH in 50 mL THF was added to a solution of 4.0 g (0.02 mol) indolyl-dimethylaminomethylketone in 30 mL THF. The mixture was stirred for 30 minutes at room temperature, followed by 2 hours on a boiling waterbath. The THF was removed via distillation and water added to the complex. By doing so, a smudgy mixture of base and aluminium hydroxide was obtained. It was extracted with acetone after decanting the water; when the acetone was dried over potassium carbonate, it was evaporated, hereby giving a brownish oil. The oil crystallized after purification with ether-petroleum ether. Recrystallization from ethyl acetate-petroleum ether gave the compound.
Yield: 2.8 g = 73.7%, mp: 48-49°C; mp of the picrate salt: 170°C.
Summary (total yields presumed) from indole:
-3 steps w/ chloro acetyl chloride:
DET: 80% x 74% x ? (40%) = 24%
DMT: 80% x ? (75%) x 49% = 29%
-4 steps w/ acetonitrile:
DMT (NaBH4): 96% x 75% x 84% x 49% = 30%
DMT (LAH): 96% x 75% x 84% x 74% = 45%
(Unrelated:) Shulgin yield from indole with oxalyl chloride / LiAlH4 route:
DMT: 79% x 91% = 57%
Well, those are viable alternative routes, considering everybee that is interested in 2C-B has already acetonitrile, nitromethane, bromine and NaBH4, the second route look good, as aquisition is only indole + SnCl4
Well, it is nice to see some cooperation on certain subjects
References (linked posts):
[1]: Post 448729 (Chimimanie: "DMT from 3-acetyl indole + 5-cyano/nitro DMT", Tryptamine Chemistry)
[2]: Post 448820 (Lego: "An improved method to obtain 3-acylindoles", Tryptamine Chemistry)
[3]: Post 451451 (GC_MS: "Reduction of indolyl-3-aminomethylketones", Tryptamine Chemistry), above post.
[4]: Post 451584 (GC_MS: "Synthesis of 1-, 2- and 3-(aminoacetyl)indoles", Tryptamine Chemistry)
(One Remarkable HyperLab Bee)
08-04-03 11:13
No 451643
some notes on the proposed synthesis
0) The above post is EDITED heavily. If you read it as it was posted, read it again, it is worth your attention.
1) Acylation step. Thanks to Lego, we have a nice procedure with SnCl4 in DCM/MeNO2. Probably it is the solvent system, that does the trick, since in PhH the same reagents resulted in red tars (Post 448729 (Chimimanie: "DMT from 3-acetyl indole + 5-cyano/nitro DMT", Tryptamine Chemistry)). The reason may be that the complex of indole with a Lewis acid is almost insoluble in PhH, and polymerizes instead of being acylated. 5-Cyanoindole is less prone to polymerization, and can be acylated successfully even in benzene.
Note that refluxing indole with chloroacetylchloride in PhMe without Lewis acid results in acylation of indolic nitrogen (Post 451584 (GC_MS: "Synthesis of 1-, 2- and 3-(aminoacetyl)indoles", Tryptamine Chemistry)). However, 3-chloroacetylindole was obtained in 48% yield when ClCH2COCl was slowly added to the mixture of indole and Py (1:1:1 molar ratio) in PhMe at 55° (Tetrahedron, 29, 971 (1973)).
2) Bromination of acetylindole. Bromine in methanol is a suspicious combination. They react with one another, especially above rt. Refluxing the mixture seems to be of no use. Let's see.
3) Halogen to amine exchange. Probably, toluene is not the best solvent to perform the reaction with Et2NH. In a polar aprotic solvent, theoretically, it should go faster and smoother. But then, PhMe is cheap, and if it works, why change it?
4) Reduction. 1-Propanol is not the most common solvent. Why didn't they try isopropyl alcohol? Maybe, isobutyl alcohol will work? Much experimentation is required.
So, let's wait for some curious bee to apply these findings...
Ostensibly motionless, the hare was trembling with excitement...
(Heavyweight Chempion(eer))
08-05-03 01:49
No 451772
Thanks to Lego, we have a nice procedure with SnCl4 in DCM/MeNO2. Probably it is the solvent system, that does the trick, since in PhH the same reagents resulted in red tars
Nitromethane is indeed a remarkable solvent for Friedel-Crafts acylations. Cheap, not too flamable and with a nice boiling point.
Bromination of acetylindole. Bromine in methanol is a suspicious combination. They react with one another, especially above rt. Refluxing the mixture seems to be of no use. Let's see.
Methanol has been used for bromination of acetophenones, propiophenones and butyrophenoens with good results when other solvents has failed.
Halogen to amine exchange. Probably, toluene is not the best solvent to perform the reaction with Et2NH. In a polar aprotic solvent, theoretically, it should go faster and smoother. But then, PhMe is cheap, and if it works, why change it?
Toluene/water and IPA/water has been used by myself with good results when aminating certain alpha-bromopropiophenones. But as you said, don't fix what isn't broken.
Reduction. 1-Propanol is not the most common solvent. Why didn't they try isopropyl alcohol? Maybe, isobutyl alcohol will work? Much experimentation is required.
I've seen this system before but failed to understand the mechanisms which makes n-PrOH better than IPA. Perhaps someone in our hive knows?
Moody as hell
(Hive Bee)
08-12-03 14:24
No 453174
Im having trouble comprehending the 2nd step in the above (4 steps to DMT) I sincerely appoligize if my lack of comprehension is something stupid.
Step 1:
indole + SnCl4 and acetonitrile => 3-acetyl-indole: Understood just fine
Step 2:
3-acetyl-indole => Indolyl-3-bromomethylketone:
15.9 g (0.1 mol) indolyl-methylketone was suspended in 50 mL methanol.
Where did the "indolyl-methylketone" come from? If in the first step, indole was changed to acetyl-indole, then why is the 2nd step reffering to indolyl-methylketone? Is this "indolyl-methylketone" the same substance as 3-acetyl-indole?
%
(Hive Bee)
08-12-03 14:48
No 453180
You should really study basic chemistry and nomenclature somewhat more... (especially before attempting an LAH reduction
Look at the structure of the molecule:
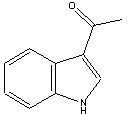
acetyl means -COCH3, so 3-acetylindole means that the acetyl group is attached to the 3-position of the indole nucleus.
ketones have the formula R-CO-R', so indolyl-methylketone is indole-CO-CH3 , a synonym for 3-acetylindole. (a more correct name would be 3-indolyl methyl ketone)
It all depends how you look at it
A Dream Within A Dream (http://www.poedecoder.com/Qrisse/works/
(Hive Bee)
08-12-03 15:39
No 453189
You should really study basic chemistry and nomenclature somewhat more
I agree, I am not educated enough by any means.
I gather by what your saying is that indolyl-methylketone is the same as 3-acetyl-indole, just another way of saying it.
Thank you for explaining.
%
(Hive Addict)
12-08-03 06:09
No 475429
(Rated as: excellent)
Patent US2814625
SPEETER, Merrill E.
Upjohn Co., 1957-11-26
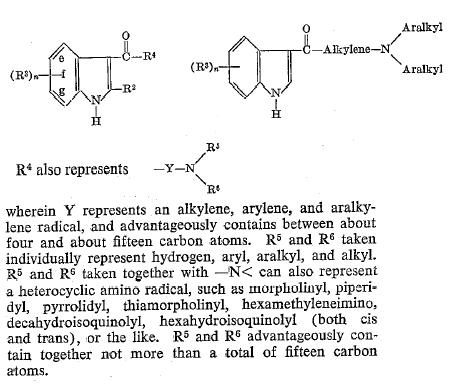
The preparation of the 3-indolyl aminohydrocarbyl ketones employed in the novel process, except in those instances wherein R3 is a cyano, carbalkoxy, carboxy or acyloxy radical, can be readily accomplished by utilizing processes well-known in the art, eg., Salway, supra, prepared a 3-indolyl aminohydrocarbyl ketone by reacting an indole with a Grignard reagent to produce an indole Grignard reagent, and reacting the thus-produced indole Grignard reagent with a haloacyl halide to produce a 3-indolyl halohydrocarbyl ketone. In some instances in addition to the desired 3-indolyl halohydrocarbyl ketone a 1,3-bis-halohydrocarbyl product may result, but the 1-halohydrocarbyl substituent is readily cleaved to produce an additional quantity of 3-indolyl halohydrocarbyl ketone.1
1'-[1'-(N,N-diethylamino)-methyl]-3-indo
Fifteen grams of 1'-(1'-chloromethyl)-3-indolyl ketone was dissolved in 200 milliliters of benzene and to this solution was added 50 grams of diethylamine. The mixture was refluxed for thirty minutes, cooled, and the cooled mixture was extracted with dilute hydrochloric acid. On addition of base to the combined extracts a solid separated which was recrystallised from isopropanol. Sixteen grams (90% yield) of 1'-[1'-(N,N-diethylamino)-methyl]-3-indo
Analysis - Calculated for C14H18N2O: C, 73.00; H, 7.87. Found: C, 72.80; H, 7.72
1'-[1'-(N,N-diethylamino)-methyl]-3-indo
Three grams of the resulting ketone was dissolved in 100 mL methyl acetate, and into this solution was added excess hydrogen chloride in isopropanol. The resulting 1'-[1'-(N,N-diethylamino)-methyl]-3-indo
Analysis - Calculated for C14H18ClN2O: C, 63.49; H, 7.18; N, 10.50. Found: C, 63.63; H, 7.26; N, 10.45.
3-[2'-(N,N-diethylamino)-ethyl]-indole
A solution of 2.3 grams of 1'-[1'-(N,N-diethylamino)-methyl]-3-indo
Analysis - Calculated for C14H21ClN2: C, 66.53; H, 8.37; N, 11.07; Cl, 14.02. Found: C, 66.29; H, 8.47; N, 10.76; Cl, 13.81.
1 : Saxton, J. Chem. Soc. 3592 (1952)
The Other War (http://www.markfiore.com/animation/drug
(Moderator)
12-16-03 15:03
No 477258
Maybe your product can be recrystallized from hexane or a hexane / ethylacetate. You probably have to experiment... BTW, If you run a reaction the first time you should do it on a way smaller scale (e.g. 500 mg).
(Chief Bee)
12-18-03 12:50
No 477681
As to avoid too high local concentrations of bromine, which could lead to increased formation of dibrominated ketone instead of monobrominated dito.
The Hive - Clandestine Chemists Without Borders
(Newbee)
12-19-03 05:23
No 477822
Anybody knows anything about the possible activity of beta-keto-tryptamines in analogy with cathinones. I guest that carbonyl would make them more liable to the methabolism but it would be worth tasting. After all the tryptamines are much more tolerant to the side chain modifications. If it works it would be a great way to eliminate the need of LAH reduction. But the diethylamine derivative should be used to make shure it would be orally active as I think the kethone would be dificult to smoke (more prone to pyrrolisis?).
“The real drug-problem is that we need more and better drugs.” – J. Ott
(Chief Bee)
01-03-04 09:27
No 480256
It appears my sample was too polar. This is confusing to me, for if I isolated this sample on a separate plate using a different solvent system, how would I be able to identify the different spots the sample may give, as I have nothing to check them against using that system??
I think you have done most of what can be done with TLC now. You have proved that your product is way more polar than the acetylindole and the bromoacetylindole (exactly what would be expected from a tertiary amine). You cannot use TLC to prove the identity of a substance, only prove that it is dissimilar to other reference compounds (exactly what would be expected if the reaction went as it was supposed to).
Now you can perform an acid/base extraction on the product, isolate the freebase and gas it with HCl gas. This should give you relatively pure amine salt (any remaining acetylindole/bromoacetylindole won't carry through). I assume that the compound is too high-boiling for a vacuum distillation to be feasible.
Am I correct that the sample is too polar or does it appear too concentrated?
The sample is too polar for your current elution system, as it barely moves from the starting line. At least #2 seems to be of proper dilution, and #1 may be okay too.
I am finding acetone disolves all these chems very nicely, where as methanol or dcm doesn't do well unless heated, is it ok to use acetone as a means of disolving your sample to be spotted?
That's fully okay, as long as you make sure that all of the acetone has evaporated before eluting the plate.
The Hive - Clandestine Chemists Without Borders
(Chief Bee)
01-04-04 09:09
No 480371
The solubility of NaBH4 is 4g/100g ethanol, so one could expect the solubility to be somewhat lower in propanol, say 2-3g/100mL - try to use enough 1-propanol for both the reagent and the reactant to dissolve. I suggest you add a concentrated 1-propanol solution of the ketone to a concentrated solution of sodium borohydride in the same solvent.
Yes, when they say "twice its weight" it is as simple as 1g:2g. Otherwise they would have written "two molar equivalents" or similar.
The Hive - Clandestine Chemists Without Borders
(Chief Bee)
01-07-04 17:20
No 481021
Publications containing syntheses of 3-(2-Dimethylaminoacetyl)-indole (a.k.a. Beta-keto-DMT)
Patent US2821532 <- Includes NaBH4 reduction, but the end product is the alcohol, not DMT...
J. Chem. Soc. 1984-1988 (1956)
J. Heterocycl. Chem.; EN; 3; 1966; 5-8.
Pharm. Chem. J. (Engl.Transl.) 10, 532-536 (1970)
Khim. Farm. Zh. 4(10), 5-9 (1970)
Arch. Pharm. Ber. Dtsch. Pharm. Ges. 294, 484-487 (1961)
Suggested recrystallization solvents (freebase, mp 208-209°C) include ethanol, isopropanol and diglyme.
The Hive - Clandestine Chemists Without Borders
(Moderator)
01-16-04 17:33
No 482931
Quaternary amines maybe?
(Moderator)
01-17-04 15:08
No 483090
Quaternary amines result from the reaction of two bromoketones with one dimethylamine resulting in a quaternary amine salt N+R4. To prevent this you have to use a large excess of dimethylamine all the time.
The reaction products (beside quaternary ammonium salts) are probably the hydrobromide of dimethylamine and of the target compound. Your product has to be freebased in order to become THF-soluble and to be used in the reduction.
The solution has to be added dropwise from a dropping funnel to the boiling reduction, otherwise you are risking an overboiling. An oilbath is better compared to a water bath because you dont want water and alanate in close proximity... The heating bath has to be hotter than the boiling points of the solvents to be refluxed (30°C or so more) because you have to pump a lot of energy into the solution for boiling and refluxing
(Stranger)
01-22-04 14:35
No 484076
Organic Chemistry second edition (Maitland Jones, 2000) says that halogenation at the alpha position of methyl ketones is accomplished by addition of X[sub]2 in acid environment. The mechanism for this substitution depends on the acid functionality: the electron density on the ketone oxygen removes the dissociable hydrogen from the acid, forming a transition state (resonance stabilised by a tertiary carbocation). The equilibrium is forced toward the enol form, in which case the double bond electron density makes a bond with X--X. The product is halogenated ketone and H-X.
The examples provided in the text use acid environments like HNO[sub]3/CH[sub]3COOH, i.e. strong acids. Since the reaction appears to depend on the dissociable proton to form the enol, yields are probably influenced by the relative acidity of the solvent system.
Methanol is not particularly acidic--pKa = 15.2. Compare that to nitric acid (-1.4) or carboxylic acids (4-5). I wonder if some of the yield problems that are being encountered result from methanol being a poor acid? Maybe using a solvent system such as the above mixture would overcome that problem.
(Chief Bee)
02-01-04 12:48
No 485868
It is always good to start hydride reductions cold, or at least keep them at room temp throughout the addition. It is not as critical as in the reductive amination procedure as there is no as unstable compounds present here as the imine in the other one.
The Hive - Clandestine Chemists Without Borders