10-25-02 03:33
No 372508
Is there really no interest in what might be a cheap and unsuspicious alternative to the anti-social chlorinators SOCl2, POCl3, POCl5 ?
Also, I dunno much about peptide chemistry, but might there not be possible application in the synth of lysergic acid amides ?
Mountain Boy
(Chief Bee)
11-22-02 04:59
No 382214
I don't think people knows that trichloroisocyanuric acid (cyanuric chloride) is available OTC as a pool chlorination agent. Either that, or people fail to realize that it can be used with GAA to make acetic anhydride (as there is no step-by-step writeup available).
Edit: The procedure in Post 383271 (Aurelius: "Cyanuric chloride: -COOH to -COCl/COOR/CONR2", Novel Discourse) can be used to convert acetic acid to acetyl chloride, and in ../rhodium /anhydri
(Hive Bee)
11-22-02 06:03
No 382228
Well it may not be that good. As far as I understand, cyanuric chloride [CAS 108-77-0] is not the same as the sodium salt of trichloroisocyanuric acid [CAS 87-90-1] used in swimming pools (somebee confirm this ?).
But even though cyanuric chloride might not be OTC, purchasing it from a chem supplier would still be unsuspicious I think.
Then GAA __> acetyl chloride __> acetic anhydride is just one of the possible uses.
Mountain Boy
(Chief Bee)
11-22-02 06:51
No 382242
(Rated as: excellent)
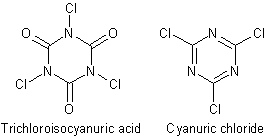
You're right, it seems like they are two separate entities, even though the similarities are striking.
Cyanuric Chloride
Synonyms: 2,4,6-Trichloro-1,3,5-triazine, Chlorotriazine, Trichlorocyanidine, Tricyanogen chloride, Cyanuric acid trichloride, 2,4,6-Trichloro-s-triazine
CAS No: [108-77-0]
Mol. Weight: 184.4
Melting point: 154°C
Boiling point: 192°C
Water Solubility: Decomposes
Synthesis of Cyanuric Chloride
Cyanuric chloride is manufacture by the reaction of chlorine with HCN, followed by trimerization of the formed cyanogen chloride.
Chlorine and HCN are added to the reactor (chlorinator) where the reaction to form cyanogen chloride (CNCl) takes place at a temperature between 20°C and 40°C. The cyanogen chloride formed in the chlorinator is washed with water in a scrubber. The cyanogen chloride, which is now devoid of HCl, is passed through a drying unit to remove traces of water. After exiting the dryer, chlorine is added to the cyanogen chloride and the mixture sent to the trimerizer where the CNCl is trimerized on activated charcoal at temperatures above 300°C to form cyanuric chloride. The cyanuric chloride vapors from the trimerizer are then condensed to form a solid product. The CNCl yield in this process exceeds 95% and the (CNCl)3 yield exceeds 90%.
Reference: http://www.epa.gov/ttn/chief/le/cyanide.
Trichloroisocyanuric Acid
Synonyms: TCCA, TCICA, ICC, Isocyanuric chloride, 1,3,5-Trichlorohexahydro-1,3,5-triazine-
CAS No: [87-90-1]
Mol. Weight: 232.44
Melting point: 225-230°C (decomposition)
Water Solubility: ∼12 g/L (20°C)
Synthesis of Trichloroisocyanuric Acid
Isocyanuric acid is manufactured by pyrolysis of urea:
3 H2NCONH2 --(200-300°C)--> C3H3N3O3 + NH3
(Urea) (Isocyanuric acid)
To obtain trichloroisocyanuric acid, isocyanuric acid is chlorinated in the presence of sodium hydroxide:
C3H3N3O3 + 3 NaOH + 3 Cl2 --> C3Cl3N3O3 + 3 H2O + 3 NaCl
(Isocyanuric acid) (trichloroisocyanuric acid)
(Hive Bee)
11-22-02 09:13
No 382283
Just in case anybody is interested, Post 381692 (SpicyBrown: "High yield aldoxime --> nitrile conversion", Novel Discourse) contains a procedure for a beckmann rearrangement on ketoximes and a conversion to nitriles from aldoximes using cyanuric chloride.
-SpicyBrown
(Chief Bee)
11-22-02 16:50
No 382428
Cyanuric Chloride can also be used as a mild alcohol -> ketone oxidant: Post 246577 (PEYOTE: "Alternative Oxidation", Chemistry Discourse)
(Hive Bee)
11-23-02 13:45
No 382653
hey Rhodium, what are the basics behind formation of the AA from GAA? how exactly does that work? aurelius specifically interested in how it would work in terms of other anhydrides (including mixed).
(Chief Bee)
11-24-02 01:48
No 382776
Well, if you have an agent capable of turning acids into acid chlorides (RCOOH -> RCOCl), then reacting that chloride with another molecule of acid (doesn't have to be the same), then you have an easy route to acid anhydrides (RCOCl + R'COOH -> RCO-O-OCR' + HCl).
(Hive Bee)
11-25-02 13:34
No 383271
(Rated as: excellent)
Cyanuric chloride: A Useful Reagent for Converting Carboxylic Acids Into Chlorides, Esters, Amides and Peptides (1)
K. Venkataraman* and D.R. Wagle
National Chemical Laboratory, Poona 8, India
Recent examples of the sustained interest in reagents for the conversion of carboxylic acids to chlorides, amides and peptides are: (a) 2,4,6-trinitrofluorobenzene for the preparation of amides and esters (2), (b) isocyanides for peptide synthesis (3), (c) oxalyl chloride and a catalytic amount of DMF for the conversion of carboxylic acids to chlorides via the t-butyldimethylsilyl esters (4), and (d) esterification of carboxylic acids in the presence of DCC and a catalytic amount of a 4-dialkylaminopyridine (5). Another useful reagent for these purposes is cyanuric chloride (CC), readily available as an intermediate for the manufacture of reactive dyes, fluorescence, brightening agents and agricultural pesticides.
Senier recorded in 1886 the preparation of acetyl and benzoyl chlorides by heating the sodium salts with CC at 100 C for 8 hours (6). Refluxing CC with a large excess of glacial acetic acid has been suggested as a method for the preparation of cyanuric acid, acetyl chloride being simultaneously formed (7). In the method now described the reaction is carried out at room temperature and CC separates as an insoluble product; the solution containing the acid chloride and any unconverted acid can be used directly for further reactions.
When CC in acetone is treated with 1 or 2 mol of carboxylic acid and 1-2 mols of triethylamine (TEA), the acid chloride is rapidly formed, presumably via the sigma adduct (I) resulting from a nucleophilic attack of RCOO(-) on CC. CC is converted into insoluble dichlorohydroxy- or chlorodihydroxy-s?-triazine (8), which have also been characterized as the corresponding dianilino or monoanilino derivatives (9).
The general procedure is to TEA (0.02mol) to a solution of the carboxylic acid (0.02mol or 0.01 mol of a dicarboxylic acid, etc.) and CC (0.01mol) in acetone (20ml or minimum volume required for clear solution) at 20-8(?)0*C. After stirring for 3hrs when no CC remains in solution, acetone is remove under reduced pressure and the acid chloride taken up in carbon tetrachloride. Alternatively, when the desired product is an ester or amide, the alcohol, phenol or amine (0.02mol) is added to the reaction mixture, which is then stirred for 2hrs. The triazine derivative is filtered off and the acetone solution worked up as usual.
Three dipeptides were prepared from (1) Z-glycine, (2) Z-l-valine and (3) boc?-l-valine. After treatment with CC and TEA, glycine ethyl ester hydrochloride is added as a suspension in acetone (10ml) and TEA (0.04mol). The reaction mixture is added to ice-water and extracted with chloroform. The dipeptide, recovered from the chloroform solution after the removal of unconverted acid and amine, is then crystallized (14,15,16). The three peptides and Z-glycine trichlorophenyl ester were characterized by their NMR and mass spectra in addition to their melting points.
The yields recorded in the Table are of the isolated products after purification by acid and base washing, when they were chromatographic…? homogenous and had mp’s about 5*C lower than the literature values, but before the crystallization; the recovery uncovered acid was not taken into account. A simple routine procedure using the regenerated TEA was followed for the preparation of esters and amides, and no attempt was made to optimize conditions for maximum yields.
We are grateful to……
Table
Acids coverted to chlorides, amides or esters
Acid amine, phenol product(10) yield
Or alcohol %
Acetic aniline anilide 84
p-aminophenol p-hydroxyacetanilide
Trifluoroacetic aniline anilide(11)
l-valine N-trifluoroacetyl-l- 50
valine(12)
Oxalic aniline anilide 52
Malonic chloride or 0
Anilide
Succinic aniline anilide 55
Z-glycine 2,4,6-trichloro- trichlorop
phenol ester (13)
Phenylacetic aniline Anilide 86
Benzoic chloride 81
Aniline anilide 87
o-phenylene- N,N-dibenzoyl-2- 68
diamine phenylenediamine
o-aminophenol N-o-dibenzoyl-2- 73
aminophenol
methanol methyl benzoate 82
p-nitrobenzoic chloride 58
Cinnamic aniline anilide 93
Aspirin methanol methyl ester 45
sorry- didn't feel like doing the refs right now
if you want them, aurelius will do them, just later.
(Hive Bee)
11-25-02 13:54
No 383284
(Rated as: good read)
http://www.heidelberg.edu/depts/chm/tria
(Chief Bee)
11-26-02 00:27
No 383427
(Rated as: excellent)
Development of a Process for Triazine-Promoted Amidation of Carboxylic Acids
Org. Proc. Res. Dev., 3 (3), 172-176, 1999.
Abstract:A process has been developed for the triazine-promoted amidation of carboxylic acids. We have identified 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride) as a cost-effective reagent for this transformation. The procedure is a suitable alternative to traditional amidation processes when an acid chloride cannot be prepared from the corresponding carboxylic acid due to safety, stability, or handling concerns.
Introduction
Amides are typically prepared by coupling an amine with an acid chloride generated from the parent carboxylic acid. While acid chlorides are highly reactive reagents, they suffer from several disadvantages. When an acid chloride is not commercially available, its manufacture can present handling and safety concerns. Though technically simple, conversion of a carboxylic acid to the corresponding acid chloride can be cumbersome in a plant. The acid chloride is often generated from an acid derivative such as an ester as part of a multistep synthesis; in this case, the carboxylic acid intermediate must be filtered and dried or extracted into a suitable solvent and dried azeotropically. These steps add cost through increased cycle times.
The typical reagents employed to prepare an acid chloride from a carboxylic acid are corrosive (thionyl chloride) or toxic (phosgene). While these reagents are inexpensive and very useful in the preparation of many acid chlorides, there are some situations in which these reagents are unsuitable. Certain carboxylic acids, such as some optically active compounds, are unstable to acidic conditions. Additionally, some smaller manufacturing facilities such as pilot plants may not have the facilities or the desire to handle toxic or highly corrosive reagents. Acid chlorides also present handling and storage issues due to their corrosivity and water reactivity.
Acid anhydrides have been utilized as alternatives to acid chlorides in amidation procedures. However, this procedure is often problematic. If the carboxylic acid is converted to a symmetric anhydride, 1 equiv of carboxylic acid is lost as the byproduct of the amidation reaction. While expensive acids may be isolated and reconverted to anhydride, the additional processing required may be costly. If the acid is sufficiently expensive, a mixed anhydride may be prepared from an inexpensive material such as acetic acid. However, it is unlikely that only one of the acid components will couple with the amine, and a mixture of amide products is often obtained.
Because of the disadvantages associated with the use of acid chlorides and anhydrides in amidation reactions, amidation of carboxylic acids is an active area of research. A common approach involves treatment of the acid with a reagent to form an activated intermediate, which is then treated with an amine in situ to form the amide product. This approach is primarily of interest for the preparation of peptides. Many reagents have been identified that allow coupling of carboxylic acids and amines. However, most are quite expensive, and separation of the byproducts produced is difficult.
We have developed a process suitable for large-scale preparation of amides using carboxylic acids and a triazine reagent as the promoter. While our interest is in the preparation of amides which have activity as agrochemicals, the work may have peptide synthesis applications as well.
Results and Discussion
Very few reagents reported to promote amidation of carboxylic acids are suitable for adaptation to large-scale manufacture. Many of the known reagents are costly, particularly those developed for the synthesis of peptides. Additionally, separation of the byproduct produced from the activating reagent is difficult unless chromatographic techniques are employed. The most economically viable reagent disclosed in the literature is 2-chloro-4,6-dimethoxy-1,3,5-triazine.1 The reagent is commercially available and is readily prepared from commercially available 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride) using methanol and aqueous base.2 When treated with equimolar amounts of a carboxylic acid and a tertiary amine base, the activated species shown in Scheme 1 is formed.3 This intermediate reacts with an amine to form the amide and an insoluble hydroxytriazine byproduct, which is readily removed by filtration.
Our investigation began with an evaluation of the reaction conditions reported in the literature for amide formation using 2-chloro-4,6-dimethoxy-1,3,5-triazine. A carboxylic acid of interest, 3,5-dichloro-4-methylbenzoic acid, was combined with the triazine reagent and N-methylmorpholine at ambient temperature. The intermediate was treated with several amines, affording the desired amides cleanly and in good yield (Table 1).
Once the dimethoxytriazine reagent was proven to be an effective amidation promoter, we set out to improve upon the reported reaction conditions to develop a process suitable for large-scale synthesis. First, we examined the utility of cyanuric chloride, the commercially available parent compound of the dimethoxytriazine reagent, as an amidation promoter. We theorized that use of the trichlorotriazine promoter would permit the use of only 0.33-0.5 mol of triazine reagent per mole of carboxylic acid employed. We found several references to the use of cyanuric chloride for amidation,4 but the procedures were not well suited to scale-up. The triazine reagent and carboxylic acid were typically employed in a 1:1 ratio, which circumvents the advantage of employing the trichlorotriazine reagent. In each case, tertiary amine bases were used to generate the activated intermediate. These conditions were undesirable from cost and waste generation considerations.
We also found that there was some disagreement in the literature regarding the product of the reaction between cyanuric chloride and a carboxylic acid. It has been proposed that the product of this reaction is the corresponding acid chloride. If this were the case, this amidation procedure would be undesirable for racemizable substrates such as amino acids due to the presence of the hydrogen chloride byproduct. However, in our hands, no acid chloride was observed after the activation step. We successfully performed the amidation using 0.33 equiv of cyanuric chloride per mole of carboxylic acid. The experimental procedure was otherwise similar to that employed when using the dimethoxytriazine derivative. Based on these results, our proposed intermediate is the triacylated triazine shown in Scheme 2.
The nature of the base used to generate the carboxylate salt was not critical. While tertiary amine bases were effective, we preferred to develop a procedure employing an inorganic base. Aqueous sodium hydroxide was suitable, and the presence of water did not adversely affect intermediate formation. It should be noted that the sodium hydroxide was depleted by adding the carboxylic acid before the triazine reagent in order to avoid hydroxytriazine formation. The intermediate formed rapidly, as evidenced by precipitate formation and heat evolution upon addition of the triazine to the carboxylate salt. We also utilized preformed carboxylate salts in the reaction, demonstrating that base is not required for this procedure. This modification is useful when the desired substrate is a commercially available carboxylate salt or when it is necessary to isolate the salt due to purification or other handling considerations. Upon addition of a primary amine, a second exotherm was observed; amidation was typically complete within 1 h. A summary of the experimental data appears in Table 2.
The cyanuric chloride-promoted process has several advantages that make it suitable for large-scale manufacture of amides. The ability to use only 0.33 equiv of the triazine promoter is advantageous because it minimizes reagent utilization and byproduct generation compared to the dimethoxytriazine procedure. The reaction is robust, as the presence of water in both the activation and amidation steps is tolerated. This is an important feature for two reasons. First, inexpensive inorganic bases may be used to generate the carboxylate anion required in the activation step. This improvement simplifies the byproduct streams; we have eliminated the amine bases used in the literature which would have to be disposed of or recycled. The water tolerance of the procedure is also desirable because some amines of interest are isolated as solutions in solvent-water azeotropes. Another advantage of the process is that separation of the byproduct is simple. The precipitated cyanuric acid is readily separated by filtration, while the amide product remains in solution. Residual cyanuric acid can be removed with a base wash. Another attractive feature of this process is that amidation occurs readily, even when sterically hindered primary tert-alkylamines are employed in the reaction.
While the yields achieved to date are acceptable (typically 65-75%), they do not currently match yields obtained from traditional amidation methods. Occasionally, unreacted carboxylic acid is observed in the reaction mixture and removed with a base wash. No side products are observed in most cases, indicating that yield losses occur during workup. We believe that the losses are occurring during filtration, when product is trapped with the cyanuric acid byproduct. These losses can be minimized by effective deliquoring and washing during solid-liquid separation of the byproduct and the solution containing the amide product.
Conclusions
The amidation of carboxylic acids promoted by triazine reagents is an alternative to traditional amidation procedures employing acid chlorides. We have developed a process for this transformation which utilizes 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride) as the activating agent. The ability to avoid the preparation and handling of acid chlorides and the ease of byproduct separation are key features of this chemistry.
Experimental Section
General Procedure for 2-Chloro-4,6-dimethoxy-1,3,5-triazine-Pr
3,5-Dichloro-4-methyl-N-(1,1,3,3-tetrame
3,5-Dichloro-N-(3-chloro-1-ethyl-1-methy
3,5-Dichloro-N-(1,1-diethyl-2-propynyl)-
General Procedure for 2,4,6-Trichloro-1,3,5-triazine-Promoted Amidation Using N-Methylmorpholine as Base. A slurry of the carboxylic acid (1 equiv) in a polar organic solvent was treated with 2,4,6-trichloro-1,3,5-triazine (0.33 equiv) and N-methylmorpholine (1.02 equiv). A 12-13 C exotherm was observed, and a precipitate formed upon addition of the amine base. After the slurry was stirred for 1 h, the primary amine (1.02-1.05 equiv) was added; a 3-5 C exotherm was observed. The reaction was judged to be complete by GC analysis. The slurry was cooled to room temperature and filtered. The solid was washed with a minimal amount of solvent. The filtrates were combined and washed with 1 M sodium hydroxide solution and with water. The organic layer was dried over sodium sulfate, and the solvent was removed by evaporation under reduced pressure. The residue was dried under vacuum to yield the amide product.
3,5-Dichloro-N-(1,1-dimethyl-2-propynyl)
N-(3,4-Dichlorophenyl)propionamide (5).10 Following the general procedure, a solution of propionic acid (10.0 g, 134.99 mmol) in n-butyl acetate (100 mL) was treated with 2,4,6-trichloro-1,3,5-triazine (8.30 g, 44.99 mmol) and N-methylmorpholine (13.93 g, 137.7 mmol). The resulting slurry was treated with 3,4-dichloroaniline (22.31 g, 137.7 mmol), and the reaction mixture was stirred at 23 C for 1 h. Workup afforded the amide as a pale tan solid (21.54 g, 73%): mp 86-88 C (lit.11 mp 86-91 C). The spectral data obtained were identical with those of an authentic sample.
3,5-Dichloro-N-(1-ethyl-1-methyl-2-propy
3,5-Dichloro-N-(1-ethyl-1-methyl-2-propy
N-(1,1,3,3-Tetramethylbutyl)benzamide (7):12 Amidation Using Carboxylate Salt without Added Base. A slurry of potassium benzoate (2.0 g, 12.48 mmol) in 40 mL of 7:1 acetonitrile-water was treated with 2,4,6-trichloro-1,3,5-triazine (0.76 g, 4.12 mmol). The mixture was stirred for 1 h, and tert-octylamine (1.69 g, 13.10 mmol) was added. A 4 C exotherm was observed. The reaction was stirred at 23 C for 1 h. Workup as described in the general procedure afforded the amide as a white solid (1.92 g, 65%): mp 63-65 C (lit.13 mp 67-69 C).
N-(1-Ethyl-1-methyl-2-propynyl)benzamide
References
1. (a) Kaminski, Z. J. Tetrahedron Lett. 1985, 26, 2901.
(b) Kaminski, Z. J. Synthesis 1987, 917.
(c) Taylor, E. C.; Schrader, T. H.; Walensky, L. D. Tetrahedron 1992, 48, 19.
2. (a) Menicagli, R.; Malanga, C.; Peluso, P. Synth. Commun. 1994, 24, 2153 and references therein.
(b) Cronin, J. S.; Ginah, F. O.; Murray, A. R.; Copp, J. D. Synth. Commun. 1996, 26, 3491.
3. Kaminski, Z. J.; Paneth, P.; Rudzinski, J. J. Org. Chem. 1998, 63, 4248.
4. (a) Venkataraman, K.; Wagle, D. R. Tetrahedron Lett. 1979, 3037.
(b) Wagle, D. R.; Garai, C.; Chiang, J.; Monteleone, M. G.; Kurys, B. E.; Strohmeyer, T. W.; Hegde, V. R.; Manhas, M. S.; Bose, A. K. J. Org. Chem. 1988, 53, 4227 and references therein.
(c) Sainsbury, M.; Strange, R. H.; Woodward, P. R.; Barsanti, P. A. Tetrahedron 1993, 49, 2065.
(d) Hinz, W.; Just, G. Can. J. Chem. 1987, 65, 1503.
5. Michelotti, E. L.; Young, D. H. Patent US5304572
6. Michelotti, E. L.; Rayle, H. L.; Stephens, R. W.; Zabrodski, W. J. Patent US5874466
7. Rayle, H. L.; Roemmele, R. C.; Stephens, R. W. Patent US5859254
8. Propyzamide, manufactured and marketed as Kerb herbicide by Rohm and Haas Co., Philadelphia, PA.
9. Swithenbank, C.; McNulty, P. J.; Viste, K. L. J. Agric. Food Chem. 1971, 19, 417.
10. Propanil, manufactured and marketed as Stam herbicide by Rohm and Haas Co., Philadelphia, PA.
11. Schaefer, W.; Wegler, R. French Patent Patent FR1339155 Chem. Abstr. 1964, 60, 2861a.
12. (a) Lacey, R. N. J. Chem. Soc. 1960, 1633.
(b) Alender, J.; Morgan, P.; Timberlake, J. J. Org. Chem. 1983, 48, 755.
13. Johnson, R. A.; Murray, H. C.; Reineke, L. M. J. Am. Chem. Soc. 1971, 93, 4872.
14. Hennion, G. F.; Teach, E. G. J. Am. Chem. Soc. 1953, 75, 1653.
(Chief Bee)
11-26-02 00:48
No 383431
(Rated as: excellent)
Trichloroisocyanuric Acid: A Safe and Efficient Oxidant
Ulf Tilstam and Hilmar Weinmann
Organic Process Research & Development 6, 384-393 (2002) (../rhodium/pdf /trichloroiso
Abstract
The literature on trichloroisocyanuric acid (TCCA) has been reviewed. TCCA is a safe and efficient reagent, useful for chlorination and oxidation even on large scale.
(Hive Bee)
11-26-02 09:36
No 383566
take look at schemes 12, 16 and 26 (especially part b- hello GBL), 14,19, 27 (part b for benzaldehyde from benzyl alcohol), all of 31, 38 (part c,formation of acid chlorides, part d, formation of cyanides from amides- dehydration reactions)
(Chief Bee)
11-26-02 10:36
No 383590
(Rated as: good read)
Reaction of 1,3,5-Trichloro-2,4,6-trioxohexahydro-s-
J. Org. Chem. 31, 3836 (1966)
In order to moderate the reaction, the reaction flask was placed in an ice bath. To 60 ml (53.28 g, 0.74 mole) of tetrahydrofuran, containing 6 ml of water, was added 23.24 g (0.10 mole) of trichloroisocyanuric acid a t such a rate as to maintain a gentle reflux. Upon addition of the first amount of trichloroisocyanuric acid, a yellow color appeared and quickly faded. A white precipitate formed immediately. After all of the trichloroisocyanuric acid was added, the reaction mixture was allowed to stir overnight. Cyanuric acid precipitated almost quantitatively and was removed by filtration. A small sample of the reaction mixture at this point when subjected to gas chromatography showed a peak at the characteristic retention time for gamma-butyrolactone. The filtrate was combined with 60 ml of ether and extracted three times with 20-ml portions of 5% aqueous sodium bicarbonate. The ether layer was distilled, and found to contain only tetrahydrofuran. The water layer was acidified with aqueous hydrochloric acid, and the water and hydrochloric acid were removed by azeotropic distillation with benzene. The benzene was removed by distillation, leaving 2.55 g (19%) of gamma-butyrolactone, bp 88-89°C/10mmHg, nd 1.4332. The infrared and nmr spectra were identical with those of an authentic sample of gamma-butyrolactone.
The reference for 1,4-Butanediol to GBL is Synth. Commun. 25, 719 (1995)
(Stranger / Eraser)
11-27-02 14:48
No 384010
Post 340852 (Captain_Mission: "GBL from THF using trichloroisocyanuric acid", Novel Discourse)
(Hive Bee)
07-30-03 06:17
No 450704
(Rated as: excellent)
Cyanuric chloride can also bee used to convert alkyl alcohols to alkyl chlorides.
These two methods has not been posted yet (according to TFSE) and are not on Rhodium's page.
The first method is for lower alcohols (like MeOH or isopropylalcohol):
J. Org. Chem., 1970, 35(11), 3967-3968 (http://www.geocities.com/legochemistry/
Experimental section
A typical preparation involves heating the alcohol (2-20 mol) to 10-20° below its boiling point and then slowly adding powdered cyanuric chloride (1 mol). A Dry Ice trap should be connected via a rubber tube to the top of the reflux condenser in order to trap the low boiling chlorides. After the addition (ca. 1-1.5 hr), the reaction mixture is cooled, filtered, and distilled. If complete conversion to the chloride is desired, excess cyanuric chloride should be added.
The second is for higher alcohols (like benzyl alcohol):
Org. Lett., 2002, 4(4), 553-555 (http://www.geocities.com/legochemistry/
DOI:10.1021/ol017168p
Representative Procedure
Chlorination of (S)-(1-Hydroxymethyl-3-methylbutyl)-carb
2,4,6-Trichloro-[1,3,5]triazine (1.83 g, 10.0 mmol) was added to DMF (2 mL), maintained at 25 °C. After the formation of a white solid, the reaction was monitored (TLC) until complete disappearance of TCT, and CH2Cl2 (25 mL) was added, followed by the alcohol (2.39 g, 9.5 mmol). After the addition, the mixture was stirred at room temperature and monitored (TLC) until completion (4 h). Water (20 mL) was added, and then the organic phase was washed with 15 mL of a saturated solution of Na2CO3, followed by 1 N HCl and brine. The organic layers were dried (Na2SO4), and the solvent evaporated to yield (S)-(1-chloromethyl-3-methylbutyl)-carba
The candle that burns twice as bright burns half as long
(Active Asperger Archivist)
07-30-03 17:01
No 450765
(Rated as: excellent)
An Efficient Route to Alkyl Chlorides from Alcohols Using the Complex TCT/DMF
Lidia De Luca, Giampaolo Giacomelli,* and Andrea Porcheddu
Org. Lett., Vol. 4, No. 4, 553-555, (2002)
Abstract:
Efficient conversion of alcohols and beta-aminoalcohols to the corresponding chlorides (and bromides) can be carried out at RT in DCM, using 2,4,6-trichloro-[1,3,5]-triazine and N,N-DMF. This procedure can also be applied to optically active carbinols.
The transformation of alcohols into the corresponding alkyl halides is one of the most studied reactions in organic synthesis, and many reagents can be usually used. Often the conversion requires elaborate reagents and quite drastic reaction conditions. Most of the methods employed utilize reagents such as thionyl chloride,1 phosphorus halides,2 phenylmethyleniminium,3 benzoxazolium,4 Vilsmeyer-Haack,5 and Viehe salts.6 In this context, the development of efficient reagents to use in mild conditions has interested organic chemists. The procedure based on the use of triphenylphosphine-carbon tetrahalides seems to meet these requirements but suffers the inconvenience of generating stoichiometric quantities of triphenylphosphine oxide as byproduct. To resolve these drawbacks, (chloro-phenylthiomethylene)dimethylammo
A search of the literature revealed that the treatment of cyanuric chloride, a very cheap reagent, with alcohols furnished the corresponding chlorides.10 The reported procedure implied heating of the mixture to 10-20*C below the boiling point of the alcohol and the use of excess cyanuric chloride for the complete conversion. Indeed, this method did not seem suitable for obtaining complex organic chlorides, such as those derived from amino alcohols. An accurate examination of the former report[sup11[/sup] showed that the treatment of the adduct formed by cyanuric chloride and dimethyl formamide with ethanol resulted in the quantitative formation of HCl and ethyl chloride.
On this basis and following our recent interest in the use of [1,3,5]-triazine derivatives in organic synthesis,12 we report a very mild, efficient, and chemoselective procedure for the quantitative conversion of alcohols into the corresponding alkyl chlorides (Scheme 1).
Scheme 1:
R-OH + TCT/DMF in DCM @ RT to give R-Cl
The procedure is based on the reaction of 2,4,6-trichloro-[1,3,5]-triazine (TCT) with DMG, followed by the addition of a DCM solution of 1mol. eq. of the alcohol. At 25*C, this system effects rapidly the quantitative conversion of the alcohols to the corresponding chlorides (Table 1), which can be recovered chemically pure after a simple aqueous workup that removes the triazine byproducts. The reaction is generally fast, requiring from 10-15 minutes to 4 hours for completion in most of the cases. Reduced rates were observed with sterically constrained alcohols, such as borneol and neopentyl alcohol. As in other cases, 2-phenylsulfanyl-1-ethanol reacts very slowly (ca. 72hours). Reaction of diols gave monochlorination using 1 mol. eq. of the diol, and the conversion to dichloride is complete only using 0.5mol. equivalent. At least with the optically active alcohols we have tested, the data collected show that the reaction occurs with inversion of configuration at the chiral center.13,14
Alkyl bromides can obtained by addition of sodium bromide and the alcohol to the TCT/DMF mixture in DCM. However, in this case, a noticeable amount of the alkyl chloride may be recovered as byproduct.15 Use of sodium iodide did not lead to the formation of alkyl iodides.16
Most interestingly, the reaction is applicable for the synthesis of N-protected beta-amino chlorides. Under the usual conditions, N-protected beta-amino alcohols are in fact converted to the corresponding chlorides, with slightly reduced rates. (Table 2); however, the reaction is complete within 4 hours. Moreover, the method is compatible with the common N-protecting groups, and no deprotection was noted even with N-Boc-protected amino acids, if working in the presence of NaHCO3.
The stereochemical results indicate the occurrence of a Sn2 reaction that may be consistent with the mechanism depicted in Scheme 2. The Vilsmeyer-Haack-type complex should add the hydroxyl group of the alcohol to form the cationic species 3; subsequent nucleophilic attack of halide ion should produce the corresponding halide.17
In conclusion, the procedure reported here is operationally simple and allows a rapid and high-yielding conversion of alcohols to the corresponding chlorides and bromides under very mild conditions. The method seems to be more convenient with respect to other reports and can be used as a valid alternative to other methods, so avoiding tedious purifications or the use of more toxic reagents.
Acknowledgements This work was financially supported by the University of Sassari (Fondi ex-60%).
Note Added after ASAP:
There was a nitrogen omitted from the second reagent above the arrow in the abstract in the version posted ASAP on 1/17/02. The print and final Web version was posted (1/23/02).
Supporting Information Available:
Physical and spectroscopic data for all unknown compounds and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
References:
(1)For a review, see: Larock, R.C. Comprehensive Organic Transformations, 2nd ed.: John Wiley & Sons: (1999), 689.
(2)Weiss, R.G.; Snyder, E.I. J. Chem. Soc. Chem. Commun., (1968), 1358; JOC, (1972), 36, 403.
(3)Fujisawa, T. Iida, S; Sato, T. Chem. Lett., (1977), 1173.
(4)Mukaiyama, T; Shoda, S. I.; Watanabe, Y. Chem. Lett., (1977), 383.
(5)Benazza, T; Uzan, R.; Beaupere, D; Demailly, G. Tetrahedron Lett., (1992), 33, 4901.
(6) Benazza, T; Uzan, R.; Beaupere, D; Demailly, G. Tetrahedron Lett., (1992), 33, 3129.
(7)Gomez, L; Gellibert, F; Wagner, A; Mioskowski, C. Tetrahedron Lett., (2000), 41, 6049.
(8)Pollastri, M; Sagal, J.F.; Chang, G. Tetrahedron Lett., (2001), 42, 2459.
(9)Ren, R.X.; Xin, Wu; J. Org. Lett., (2001), 3, 3027.
(10)Sandler, S.R.; JOC, (1970), 35, 3967.
(11)Gold, H. Agnew. Chem., (1960), 72, 956.
(12)(a) Falorni, M; Porcheddu, A; Taddei, M. Tetrahedron Lett., (1999), 40, 4395.
(b)Falorni, M. Giacomelli, G.; Porcheddu, A.; Taddei, M. JOC, (1999), 64, 8962.
(c)Falchi, A; Giacomelli, G; Porcheddu, A; Taddei, M. Synlett, (2000), 275.
(d)De Luca, L; Giacomelli, G.; Porcheddu, A; Taddei, M. JOC, (2001), 66, 2534.
(e)De Luca, L; Giacomelli, G.; Porcheddu, A; Org. Lett., (2001), 3, 1519.
(f)De Luca, L; Giacomelli, G.; Porcheddu, A;Org. Lett., (2001), 3, 3041.
(g)De Luca, L; Giacomelli, G.; Porcheddu, A; JOC, (2001), 66, 7907.
(13)Giacomelli, G.; Lardicci, L. JOC, (1981), 46, 3116.
(14)Kwart, H.; Givens, E.N.; Collins, C.J. JACS, (1969), 91, 5532.
(15)The bromide can be recovered by accurate distillation or flash chromatography.
(16)No result was obtained even with the addition of tetrabutylammonium iodide. The presence of tetrabutylammonium bromide causes the formation of the alkyl bromide in low yields.
(17)Representative Procedure: Chlorination of (S)-(1-hydroxymethyl-3-methylbutyl)-carb
2,4,6-trichloro-[1,3,5]-triazine (1.83g, 10mmol) was added to DMF (2ml), maintained at 25*C. After the formation of a white solid, the reaction was monitored (TLC) until complete disappearance of TCT, and DCM (25ml) was added, followed by the alcohol (2.39g, 9.5mmol). After the addition, the completion (4hours). Water (20ml) was added, and then the organic phase was washed with 15ml of a saturated solution of sodium carbonate, followed by 1N HCl and brine. The organic layers were dried with sodium sulfate, and the solvent evaporated to yield (S)-(1-chloromethyl-3-methylbutyl)-carba
Typical yields for R-Cl is 95%+, and for R-Br, yields are 70%+ Many examples are found in the tables. (not shown in this post)
Act quickly or not at all.
(Chief Bee)
11-20-03 14:24
No 472073
(Rated as: good read)
G.A. Hiegel and M. Nalbandy, "The Oxidation of Alcohols to Ketones Using Trichloroisocyanuric Acid", Synth. Commun., 22, 1589-1595 (1992).
G.A. Hiegel, "Trichloroisocyanuric Acid", in Encyclopedia of Reagents for Organic Synthesis, L. A. Paquette, Ed., John Wiley & Sons, Ltd., Chichester, West Sussex, England, Vol. 7, p. 5072-5073 (1995).
G.A. Hiegel, Christopher D. Bayne, Yariv Donde, Gerald S. Tamashiro, and Lisa A. Hilberath, "The Oxidation of Aldehydes to Methyl Esters Using Trichloroisocyanuric Acid", Synth. Commun., 26 (14) 2633-2639 (1996).
G.A. Hiegel and Afshin K. Chaharmohal, "The TCICA Test for Distinguishing Primary and Secondary Alcohols", J. Chem. Ed., 74 (4), 423 (1997).
G.A. Hiegel, Jenny Ramirez, and Robert K. Barr, "Chlorine Substitution Reactions Using Trichloroisocyanuric Acid with Triphenylphosphine", Synth. Commun., 29 (8), 1415-14-19 (1999).
G.A. Hiegel, Christine Juska, and Michelle Kim, "The TCICA Test for Distinguishing Aldehydes and Ketones", J. Chem. Ed., 78 (8), 1105-1106 (2001).
G.A. Hiegel and Mark Rubino, "Conversion of Alcohols into Alkyl Halides Using Trichloroisocyanuric Acid with Triphenylphosphine", Synth. Commun., 32 (17), 2691 (2002).
Most Recent Article by G.A.Hiegel
 (08-10-04):
(08-10-04):Preparation of Alkyl Nitrates, Nitrites, and Thiocyanates from Alcohols Utilizing Trichloroisocyanuric Acid with Triphenylphosphine
Gene A. Hiegel; Jeremiah Nguyen; Yan Zhou, Synth. Commun. 34, 2507-2511 (2004)
DOI:10.1081/SCC-120006034
Abstract
Alcohols in acetonitrile are converted into alkyl nitrates, nitrites, or thiocyanates by the action of triphenylphosphine and trichloroisocyanuric acid along with silver nitrate, silver nitrite, or sodium thiocyanate, respectively.
(Hive Bee)
07-01-04 14:25
No 516786
I wonder if the chlorination of Phenylalinol, with TCT complex, on unprotected -NH2 will get the -OH chlorinated , since the procedure is mild reagent and may not affect the -NH2. However maybe mild is not the answer since someone stated Post 516709 (Lilienthal: "... nothing stops it but strongly acidic ...", Serious Chemistry) that perhaps a more acidic environment would help avoiding . .....
the problem is that any molecule containing a primary alkyl halide and a free NH function is extremely dimerization/polymerization-prone - nothing stops the RCH2-X part of one molecule from adding to the HNR'R" of another, irreversibly forming the corresponding alkylated amine RCH2-NR'R"
.........as posted Post 516672 (Rhodium: "It is much simpler - hoffmann alkylation:", Serious Chemistry)
Although this mechanism doesn't rely on strong acidic conditions to work but rather a "chemoselective procedure" . As in other applications* of this compound TCT has been used to convert amines to amides , but then they also used N-methylmorpholine or by using the compund also made from TCT known as DMT-MM. see...
Post 430441 (Lego: "Reduction of carboxylic acids to aldehydes", Novel Discourse)
Maybe the worse that can happen is the conversion of the -NH2 to an amide, very unlikely, or a NHCl which can be reduced along with the chlorinated OH .............java
Edit by java Rhodium do you have any of the Hiegel references i.e.
G.A. Hiegel and Mark Rubino, "Conversion of Alcohols into Alkyl Halides Using Trichloroisocyanuric Acid with Triphenylphosphine", Synth. Commun., 32 (17), 2691 (2002).
It would be interesting to read if made availble.......thanks
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/
(Hive Bee)
07-03-04 05:57
No 517157
(Rated as: excellent)
Conversion of Alcohols into Alkyl Chlorides using Trichloroisocyanuric Acid with Triphenylphosphine
Gene A. Hiegel, Mark Rubino
Synthetic Communications 32(17), 2691-2694(2002)
Abstract
Trichloroisocyanuric acid with triphenylphosphine in anhydrous acetonitrile will convert alcohols into alkyl halides.
Introduction
A variety of reagents can be used to convert alcohols into alkyl chlorides.1 Many of these reagents are water reactive and do not store well. Recently we reported that a freshly prepared mixture of trichloroisocyanuric acid (1) [1,3,5-trichloro-1,3,5-triazine-2,4,6-(1
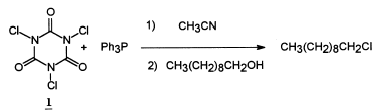
The triphenylphosphine oxide produced in these reaction can be difficult to separate from the products, however. In order to accomplish this separation, all of the reaction reported in our previous paper were carried out on a small scale with purification by means of flash chromatography. Now we report the results of reactions carried out on an 8g scale with isolation by means of extraction with pentane, which effectively separated the triphenylphosphine oxide, and purification by distillation. After an initial examination of the reaction between 1 and Ph3P, it was determined that with a 1.05 : 1 equivalent ratio of 1 : Ph3P neither reagent would be present in excess. Since there are three equivalents per mole of 1, the corresponding mole ratio is 0.35 : 1. In the general reaction procedure 1.50 equivalents of Ph3P was dissolved in anhydrous acetonitrile, and 1.58 equivalents of 1 was added slowly because the reaction is highly exothermic. The yellow mixture was then heated in a 60°C oil bath, 1.0 equivalent of the alcohol was added, and the mixture stirred for 2 h. Two different work-up procedures were used. For compounds which did not contain an aromatic ring, a large amount of water was added to the reaction mixture, the solid (cyanuric acid) was removed by filtration, the filtrate was then extracted with pentane, the pentane solution dried, and the product distilled. This procedure was not satisfactory for compounds with an aromatic ring. In those cases, the reaction was quenched with a small amount of water, and after filtration, most of the solvent was removed with a rotary evaporator. The residue was extracted with pentane, the pentane solution dried, and the product distilled. Primary alcohols gave the best yields and secondary and benzyl alcohols gave lower yields of chlorides. Cyclohexanol gave a low yield of cyclohexene. The results are summarized in Table 1.
Table 1. Conversion of Alcohols into Alkyl Chlorides using Trichloroisocyanuric acid and Triphenylphosphine
| Alcohol | Chloride | Boiling point [°C] (pres.) | Dist. yield [%] | GC purity [%] |
| 1-hexanol | 1-chlorohexane | 133.4-134.3 | 62 | 97.8 |
| 1-octanol | 1-chlorooctane | 180.7-181.4 | 65 | 99.1 |
| 1-decanol | 1-chlorodecane | 92.0-93.4 (10 torr) | 74 | 99.4 |
| 2-octanol | 2-chlorooctane | 168.2-171.8 | 52 | 96.7a |
| Cyclopentanol | Chlorocyclopentane | 110.8-112.0 | 29 | 93.5b |
| Cyclohexanol | Cyclohexene | 81.1-81.3 | 19 | 92.3c |
| Cycloheptanol | Chlorocycloheptane | 171.5-172.0 | 47 | 88.1d |
| Benzyl alcohol | Benzyl chloride | 72.8-73.2 (23 torr) | 55 | 99.3 |
| 3-phenyl-1-propanol | 1-chloro-3-phenylpropane | 85.8-88.1 (10 torr) | 70 | 99.7 |
| Cinnamyl alcohol | Cinnamyl chloride | 107.3-109.0 (10 torr) | 27 | 94.3e |
| 4-methoxybenzyl alcohol | 4-methoxybenzylchloride | 100.8-103.0 (10 torr) | 17 | decf |
aContains 1.5% 2-octene by GC;
bCOntains 4.1% pentane by GC;
cContains 6.9% pentane by GC;
dContains 10.6% of an unidentified compound;
eContains 2.6% of an unidentified compound;
fSample appeared to decompose on GC.
Both 1 and Ph3P are stable and inexpensive, but when combined, they form a reactive complex capable of converting alcohols to the corresponding alkyl chlorides.
All alcohols were of 97% or greater purity and were used as received except for benzyl alcohol and cyclohexanol which were distilled before use. Alkyl chloride and alkene standards were commercially available except for 2-chlorooctane which was prepared from 2-octanol using thionyl chloride. Anhydrous acetonitrile was used for all reactions and was obtained from Aldrich. Trichloroisocyanuric acid (1) (99% pure) was obtained from Chem Lab Products. Pentane was distilled prior to use. IR spectra were recorded using a Perkin Elmer 1650 FT-IR spectrometer. NMR were recorded using a Bruker AC-200 spectrometer. GC analyses were performed with a Hewlett Packard 5890 Series II instrument with a 6 ft. by 1/8 in. 10% Carbowax 20 M column. All reaction products were compared with standards by means of IR, NMR, and GC.
Preparation of 1-chlorodecane
To a 300-mL 3-neck RB flask were added a magnetic stir bar, a reflux condenser, and 100 mL acetonitrile by syringe. Triphenylphosphine, 19.88g (75.79 mmol), was added through a funnel, and 10 mL of acetonitrile was used to rinse the funnel. While stirring, 6.17 g (26.55 mmol) of 1 was added over about a 10 min period through a funnel and then 10 mL of acetonitrile was used to rinse the funnel. The flask was placed in a 60°C oil bath, 7.98g (50.41 mole) of 1-decanol was added to the mixture by syringe, and the reaction was stirred for 2 h. Then 30 mL water was added, the mixture filtered, and the solid washed with pentane. The filtrate was transferred to a separatory funnel and extracted with pentane (60 mL and 3×40 mL). The combined pentane solution was washed with sat. NaCl solution (50 mL) and dried over MgSO4. After filtration and removal of the pentane, the residue was distilled to give 6.61g (74%, 99.4% pure) of 1-chlorodecane. The GC retention time and the IR and NMR spectra were identical to those of the standard.
References
[1] Larock R.C. Comprehensive Organic Transformations - A Guide to Functional Group Preparations, 2nd Ed., John Wiley & Sons, Inc., New York, 1999, pp. 690–693.
[2] Hiegel G.A., Ramirez J., Barr R.K., Synth. Commun., 29 (8) , (1999) 1415.
(Hive Bee)
07-03-04 14:57
No 517157
(Rated as: excellent)
Conversion of Alcohols into Alkyl Chlorides using Trichloroisocyanuric Acid with Triphenylphosphine
Gene A. Hiegel, Mark Rubino
Synthetic Communications 32(17), 2691-2694(2002)
Abstract
Trichloroisocyanuric acid with triphenylphosphine in anhydrous acetonitrile will convert alcohols into alkyl halides.
Introduction
A variety of reagents can be used to convert alcohols into alkyl chlorides.1 Many of these reagents are water reactive and do not store well. Recently we reported that a freshly prepared mixture of trichloroisocyanuric acid (1) [1,3,5-trichloro-1,3,5-triazine-2,4,6-(1
The triphenylphosphine oxide produced in these reaction can be difficult to separate from the products, however. In order to accomplish this separation, all of the reaction reported in our previous paper were carried out on a small scale with purification by means of flash chromatography. Now we report the results of reactions carried out on an 8g scale with isolation by means of extraction with pentane, which effectively separated the triphenylphosphine oxide, and purification by distillation. After an initial examination of the reaction between 1 and Ph3P, it was determined that with a 1.05 : 1 equivalent ratio of 1 : Ph3P neither reagent would be present in excess. Since there are three equivalents per mole of 1, the corresponding mole ratio is 0.35 : 1. In the general reaction procedure 1.50 equivalents of Ph3P was dissolved in anhydrous acetonitrile, and 1.58 equivalents of 1 was added slowly because the reaction is highly exothermic. The yellow mixture was then heated in a 60°C oil bath, 1.0 equivalent of the alcohol was added, and the mixture stirred for 2 h. Two different work-up procedures were used. For compounds which did not contain an aromatic ring, a large amount of water was added to the reaction mixture, the solid (cyanuric acid) was removed by filtration, the filtrate was then extracted with pentane, the pentane solution dried, and the product distilled. This procedure was not satisfactory for compounds with an aromatic ring. In those cases, the reaction was quenched with a small amount of water, and after filtration, most of the solvent was removed with a rotary evaporator. The residue was extracted with pentane, the pentane solution dried, and the product distilled. Primary alcohols gave the best yields and secondary and benzyl alcohols gave lower yields of chlorides. Cyclohexanol gave a low yield of cyclohexene. The results are summarized in Table 1.
Table 1. Conversion of Alcohols into Alkyl Chlorides using Trichloroisocyanuric acid and Triphenylphosphine
| Alcohol | Chloride | Boiling point [°C] (pres.) | Dist. yield [%] | GC purity [%] |
| 1-hexanol | 1-chlorohexane | 133.4-134.3 | 62 | 97.8 |
| 1-octanol | 1-chlorooctane | 180.7-181.4 | 65 | 99.1 |
| 1-decanol | 1-chlorodecane | 92.0-93.4 (10 torr) | 74 | 99.4 |
| 2-octanol | 2-chlorooctane | 168.2-171.8 | 52 | 96.7a |
| Cyclopentanol | Chlorocyclopentane | 110.8-112.0 | 29 | 93.5b |
| Cyclohexanol | Cyclohexene | 81.1-81.3 | 19 | 92.3c |
| Cycloheptanol | Chlorocycloheptane | 171.5-172.0 | 47 | 88.1d |
| Benzyl alcohol | Benzyl chloride | 72.8-73.2 (23 torr) | 55 | 99.3 |
| 3-phenyl-1-propanol | 1-chloro-3-phenylpropane | 85.8-88.1 (10 torr) | 70 | 99.7 |
| Cinnamyl alcohol | Cinnamyl chloride | 107.3-109.0 (10 torr) | 27 | 94.3e |
| 4-methoxybenzyl alcohol | 4-methoxybenzylchloride | 100.8-103.0 (10 torr) | 17 | decf |
aContains 1.5% 2-octene by GC;
bCOntains 4.1% pentane by GC;
cContains 6.9% pentane by GC;
dContains 10.6% of an unidentified compound;
eContains 2.6% of an unidentified compound;
fSample appeared to decompose on GC.
Both 1 and Ph3P are stable and inexpensive, but when combined, they form a reactive complex capable of converting alcohols to the corresponding alkyl chlorides.
All alcohols were of 97% or greater purity and were used as received except for benzyl alcohol and cyclohexanol which were distilled before use. Alkyl chloride and alkene standards were commercially available except for 2-chlorooctane which was prepared from 2-octanol using thionyl chloride. Anhydrous acetonitrile was used for all reactions and was obtained from Aldrich. Trichloroisocyanuric acid (1) (99% pure) was obtained from Chem Lab Products. Pentane was distilled prior to use. IR spectra were recorded using a Perkin Elmer 1650 FT-IR spectrometer. NMR were recorded using a Bruker AC-200 spectrometer. GC analyses were performed with a Hewlett Packard 5890 Series II instrument with a 6 ft. by 1/8 in. 10% Carbowax 20 M column. All reaction products were compared with standards by means of IR, NMR, and GC.
Preparation of 1-chlorodecane
To a 300-mL 3-neck RB flask were added a magnetic stir bar, a reflux condenser, and 100 mL acetonitrile by syringe. Triphenylphosphine, 19.88g (75.79 mmol), was added through a funnel, and 10 mL of acetonitrile was used to rinse the funnel. While stirring, 6.17 g (26.55 mmol) of 1 was added over about a 10 min period through a funnel and then 10 mL of acetonitrile was used to rinse the funnel. The flask was placed in a 60°C oil bath, 7.98g (50.41 mole) of 1-decanol was added to the mixture by syringe, and the reaction was stirred for 2 h. Then 30 mL water was added, the mixture filtered, and the solid washed with pentane. The filtrate was transferred to a separatory funnel and extracted with pentane (60 mL and 3×40 mL). The combined pentane solution was washed with sat. NaCl solution (50 mL) and dried over MgSO4. After filtration and removal of the pentane, the residue was distilled to give 6.61g (74%, 99.4% pure) of 1-chlorodecane. The GC retention time and the IR and NMR spectra were identical to those of the standard.
References
[1] Larock R.C. Comprehensive Organic Transformations - A Guide to Functional Group Preparations, 2nd Ed., John Wiley & Sons, Inc., New York, 1999, pp. 690–693.
[2] Hiegel G.A., Ramirez J., Barr R.K., Synth. Commun., 29 (8) , (1999) 1415.
(Chief Bee)
09-14-04 22:03
No 531319
(Rated as: good read)
Full text of the article in Post 383271 (Aurelius: "Cyanuric chloride: -COOH to -COCl/COOR/CONR2", Novel Discourse)
Cyanuric Chloride:
A Useful Reagent for Converting Carboxylic Acids into Chlorides, Esters, Amides and Peptides
K. Venkatamaran & D. R. Wagle
Tet. Lett., No. 32, 3037-3040 (1979) (../rhodium /cyanur
The Hive - Clandestine Chemists Without Borders