02-10-03 09:35
No 406227
Zinc/Hydrazinium Monoformate Reduction of Nitro Compounds to Amines
Syn. Comm., Vol 33, No 2, pp 281-289 (2003)
Abstract:
The nitro group in aliphatic and aromatic nitro compounds also containing reducible substituents such as ethene, nitrile, acid, phenol, halogen, ester, etc., are selectively and rapidly reduced at room temperature to corresponding amines in good yields by employing hydrazinium monoformate, in the presence of commercial zinc dust. It was observed that, hydrazinium monoformate is more effective than hydrazine or formic acid and reduction of nitro group occurs without hydrogenolysis in the low cost zinc dust compared to expensive metals like palladium.
../rhodium /nitro2a
(Hive Bee)
02-10-03 10:47
No 406247
If they keep finding so many good applications for Zn, I predict it will become as expensive as Pd itself
Rh, if you have access to the journal where the article comes from, could you please have a peek at ref 29 (Friedel-Crafts) and tell us if contains anything interesting regarding our beloved syntheses?
Abusus non tollit usum
(Chief Bee)
02-10-03 11:24
No 406260
Sorry, the online edition only covers 2000-2003. There are a huge amount of interesting refs in that article though.
(Hive Bee)
02-10-03 11:30
No 406264
No problem, I'll try a new experience tomorrow: the effects of mescaline on the performance abilities of an adult male, part 1 - looking for refs and articles in a library containing 10.000s of books
Abusus non tollit usum
(Hive Bee)
02-11-03 14:29
No 406784
Damn, although I was/am on M, I was unable to locate Synth Comm in our library. Not because my synapses were bombarded with things that weren't really there, but because their subscription to the journal expired and don't want to resubscribe. Aargh! Fucking morons... They still have subscriptions on computer programming languages from the 1950s, but they lack the journals necessary to become a Ph D in Amphetaminology. Soit, I'll bug the I-order-your-articles-librarian tomorrow. I found one interesting article already: Studies on the mechanism of transfer hydrogenation of nitroarenes by formate salts catalyzed by Pd/C (JOC 56 Post 1991 (not existing) 4481-4486). They study the effects catalyst amount, stirring rate, etc on the reaction. More something for "Serious Chemistry" if you ask me, or in other words, rather interesting read if you are (frequently) involved in performing Pd/C related CTHs. Anyway, I'm not spending any more time here, the visions are calling me, have to leave!
Abusus non tollit usum
(Hive Bee)
02-12-03 07:55
No 407054
I had a look for that 'Zinc promoted Friedel–Crafts acylation' article yesterday, but couldn't find it! My library has all the Synthetic Communications journals, but I couldn't find '28 (12), (1999) 2203–2206'. Pages 2203-2206 from 1999 had no relation to the topic referenced above at all
(Chief Bee)
02-12-03 11:02
No 407116
The alcohol -> azide -> amine reaction, does it involve DEAD/PPh3?
(Hive Bee)
02-12-03 11:36
No 407127
Indeed it does Rhod: certainly triphenylphosphine, but I didn't read the article very thoroughly so I'm not sure about DEAD (what a lovely acronym). The first step involved (at least) sodium azide and 1eq. PPh3 to form the azide, then adding at least 1 more equivalent of PPh3 to reduce to the primary amine. I'm not sure about the yields, but if you're still interested I can check within a couple of days.
(Chief Bee)
02-12-03 13:00
No 407154
If you didn't bring the article with you, there is no need for you to retrieve it again. While the transformation is very interesting, DEAD is too expensive for the preparation to be of immediate intrerest.
(Hive Bee)
02-13-03 16:05
No 407537
Well the true ref was not '28 (12), (1999) 2203–2206' but '28 (12), (1998) 2203–2206' you didn't look the vol number did you?
Otherwise GREAT find your ref Syn. Comm. 30 (12) 2222-2237 (2000)
Well rhodi it doesn't use DEAD
If nobody want to post it I will post it.
(Chief Bee)
02-13-03 16:25
No 407546
(Rated as: good idea!)
That sounds very interesting, give us the article!
R-OH + CCl4 + PPh3
 R-Cl + CHCl3 + O=PPh3
R-Cl + CHCl3 + O=PPh3R-Cl + NaN3
R-N3 + NaClR-N3 + PPh3 + H2O
R-NH2 + N2 + O=PPh3- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
R-OH + CCl4 + NaN3 + 2 PPh3 + H2O
R-NH2 + CHCl3 + NaCl + N2 + 2 O=PPh3Perhaps the DMF even can be skipped by introducing a PTC into the equation, as long as the CCl4/PPh3 reaction is not water-sensitive.
(Hive Bee)
02-13-03 17:38
No 407575
(Rated as: excellent)
A new novel and practical one pot methodology for conversion of alcohols to amines G. Vidya Sagar Reddy, G. Venkat Rao, R.V.K. Subramanyan and D.S. Iyengar Synth Commun 30(12) 2233-2237 (2000)
Abstract: A convenient and efficient one pot sequence has been developed for the transformation of alcohols to amines using sodium azide, triphenylphosphine in CCl4-DMF.
(...)
Transformation of alcohols to amines is an important reaction for the synthesis of a variety of organic compounds. The most common approach for their preparation involves a three steps protocol: a) conversion of alcohols to corresponding halides or sulfonates, b) nucleophilic substitution by azide anion [2] and c) reduction of azide to amine by using various reagents [3].
Alternatively they can be prepared by a two step methodology : a) conversion of alcohol to azide by Mitsunobu reaction using hydrazoic acid, triphenylphosphine and diethylazodicarboxylate (DEAD) [4], b) reduction of azide to amine.
Although these methods works well, extra time is required to isolate intermediate products and leading to low overall yields, and involves the risk of handling explosive azides. In view of this, there is a need to develop one pot sequence. Altough there is a report of one pot method [5] involving the combination of Mitsunobu and Staudinger reactions, it is less attractive as it involves the usage of toxic and expensive reagents like HN3
Treatment of alcohols with NaN3 and two equivalents of PPh3 in CCl4-DMF(1:4) at 90°C afforded amines in an excellent yields (85-95%). Formation of amines may be visualized as the initial azide formed would react with second equivalent of Ph3P giving the iminophosphorane which in turn converted to the amine upon treatment with water. Treatment of alcohols with one molar equivalent of Ph3P afforded azides exclusively in good yields confirming the azide intermediacy. This reaction has general applicability, the results obtained with various primary and secondary alcohols are summarized in table-1. The reaction of primary alcohols was completed within 4-6 hrs, whereas secondary alcohols required longer times (8-10 hrs). All the compounds were characterised and found to be in accordance with authentic samples.
Experimental:
Typical procedure: A mixture of alcohol (2 mmol), sodium azide (2.4 mmol) and PPh3 (4.2 mmol) in 10 ml of CCl4-DMF(1:4) was warmed at 90°C with stirring. After total disappearance of starting material (monitored by TLC), reaction mixture was brought to RT and quenched by adding 5 ml of water. After stirring for 10 min., reaction mixture was diluted with ether (25 ml) and washed thoroughly with water. By trituration of ether fraction at 0°C, triphenylphosphineoxide was crystallized out and ether was filtered off. Dried over anhydrous Na2SO4, filtration and concentration of solvent afforded amines almost in pure form, which were passed through a short pad of silica gel to give pure amines.
In summary, we developed a facile and efficient one pot methodology for the conversion of alcohols to azides/amines by using readily available, cheap reagents. The major advantages of the present work are neutral reaction conditions and can be used for acid and base sensitive substrates, avoids multisteps and hazardous reagents, and offers a practical alternative to the earlier methodologies.
EDIT:Chimimanie's voice:
here they are, let me the time to type them rhodi!
References:
[1] E.C.B. Barbara, in: The chemistry of the Amino Group, Patai, S. (ed.), Wiley-interscience, London, 1968, chap 6
[2] Rolla, F.J., Org. Chem. 1982; 47 4327
[3] Suzuki, H, Takaoka, K, Chem lett 1984 1733
[4] Loibner, H, Zbiral, E, Helv Chim Acta 1976 59 2100
[5] Fabiano, E, Golding, B.T., Sadeghi, M.M., Synthesis 1987 190
Chimimanie's voice:
All in all a great procedure, my only wish is some cutoff on solvents volume. I predict great yields in that reaction scheme: 1,4dmb-> br-2,5-dmb --Mg; ethylene oxyde--> 2,5 dimethoxy phenethyl alcohol --this reaction, 90%--> 2C-H
Well thats a QUICK rating chief
(Chief Bee)
02-13-03 17:41
No 407576
Yes, I guessed the rxn mechanism!
Where are the references you are pointing to in the document?
(Chief Bee)
02-14-03 13:24
No 407896
Now available at ../rhodium /alcohol
Did they try the method on any phenethyl alcohol-like substrates? And where should I place Ref 1?
(Hive Bee)
02-14-03 13:57
No 407912
Ref 1 should be in (...)
This is (...) : Amines are a versatile class of compounds used frequently in organic synthesis, especially in the construction of heterocyclic compound [1]. <-------
Therefore, transformation of .... look above
No, they try it on benzyl alcohol(95%, 1h), on cinnamyl alcohol (92%, 2h), on n-octanol (96%, 1h), on various primary alcohol (90-95% 5h max), on steroid (85%, 7h) and on another secondary alcohol (on a cyclopropane ring, 94% 2h and 88% 7h, same compound)
(Chief Bee)
02-26-03 14:05
No 412043
(Rated as: excellent)
The above method isn't as novel as they want it to seem - the idea is over a decade old:
A Facile, One-pot Conversion of Primary Alcohols to Amines
J. Chem. Research (S), 1989, 296-297
Although primary amines can be obtained from the corresponding alkyl halides by a variety of methods, only a few of these1 allow these compounds to be prepared directly from alcohols by a one-pot procedure. Recent approaches to this problem1,2 based on the Mitsunobu reaction, suffer from commercially inaccessible reagents and/or rather tedious work-ups. The method now reported judiciously combines several known reactions.
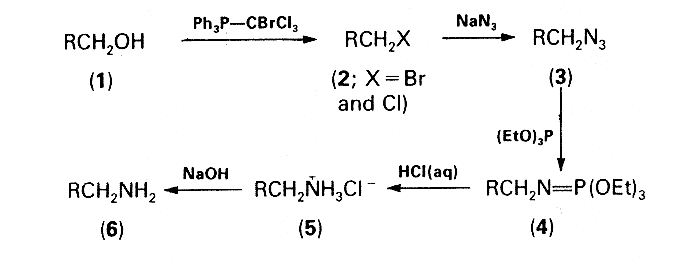
Having convincingly established the utility of transforming alkyl bromides into amines by a one-pot procedure involving the Staudinger reaction3, attention was focused upon a conversion of alcohols into bromides and/or chlorides which would then be suitable for use in this reaction without the need for isolation. Treatment of the alcohols with triphenylphosphine-tetrahalogenomethane was chosen as a reasonable approach to this problem, the reagent system triphenylphosphine-carbon tetrachloride having previously been proved very versatile for the conversion of alcohols into alkyl chlorides4. However, the relatively low reactivity of chlorides in comparison with bromides in the subsequent azidation step was discouraging. Replacement of carbon tetrachloride by bromotrichloromethane seemed a feasible method for increasing thc proportion of the more reactive bromides in the halogenated material. Moreover, in this new three-component system, Ph3P-CBrCl3 ROH, the formation of undesirable side-products, i.e. dichloromethylenephosphorane, chloromethylenephosphorane, and chloromethyltriphenylphosphonium chloride, should be less than in the Appel system5 which involves only carbon tetrachloride. The combination of triphenylphosphine and bromotrichloromethane, but with different stoichiometry, has been successfully used for the preparation of dichloromethylidenephosphoranes6. The third conceptually promising possibility of converting alcohols into the corresponding alkyl bromides by means of the two-component system carbon tetrabromide-triphenylphosphine was disqualified because of the formation of bromoform which on subsequent azidation could possibly afford highly explosive gem-diazides7 as undesirable side-products.
The effectiveness of an equimolar mixture of triphenylphosphine and bromotrichloromethane in the first step of the devised one-pot sequence depicted in the Scheme was corroborated experimentally. The mixtures of the alkyl bromides and chlorides (2) formed by the action of this reagent system on the primary alcohols (1) were then subjected to azidation and subsequent Staudinger reaction with triethyl phosphite according to the previously described procedure3. The crude iminophosphorane intermediates (4) were hydrolysed by refluxing with 20% hydrochloric acid and the solutions of the amine hydrochlorides (5) were made alkaline with sodium hydroxide. The free amines (6) were formed in good overall yields (60-70%) and characterized as their crystalline ammonium toluene-p-sulphonates. Attempts at using this procedure for converting secondary alcohols into the corresponding amines were unsuccessful: the amines were formed in very low yields (e.g. cyclohexylamine, 11%; 3-aminohexane, 21.5%) even when the azidation time was prolonged to 12 h and the amount of tetrabutylammonium bromide was increased to 10 mol%.
Experimental
The alcohols were commercially available or prepared by routine methods.
General Procedure.
The mixture of primary alcohols (1) (0.03 mol), triphenylphosphine (8.65 g, 0.033 mol), bromotrichloromethane (6.54 g, 0.033 mol), and benzene (10 ml) was refluxed gently with stirring for 2 h. It was then cooled to room temperature and, after addition of sodium azide (3.9 g, 0.06 mol), tetrabutylammonium bromide (0.48 g, 5 mol %), and dimethylformamide (10 ml), refluxed again with stirring for 6 h. The resultant mixture was then poured into water (100 ml) and extracted with benzene (2x10 ml). The organic phase was dried (MgSO4) and treated with triethyl phosphite (5.0 g, 0.03 mol) at 25-30°C for 2 h. After this solution had been stood overnight at room temperature, 20% hydrochloric acid was added and the mixture was refluxed with stirring for 2 h.
The free amine was isolated and purified by one of the following variants.
Variant A - for low-molecular-weight, water-insoluble amines
The aqueous phase was separated, made strongly alkaline with solid sodium hydroxide, and steam-distilled. The distillate was salted out, extracted with diethyl ether (4x15 ml), and evaporated using a short Vigreux column to avoid undesirable losses of amine.
Variant B - for water-soluble amines
The aqueous phase was separated, decolorized with charcoal, made strongly alkaline with solid sodium hydroxide, and extracted with diethyl ether (5x15 ml). The extract was dried (Na2SO4) and evaporated.
Variant C - for higher-molecular-weight, water-insoluble amines
Benzene was evaporated from the hydrolysate and the volatile impurities were steam-distilled from the acidic solution. The solution was made strongly alkaline with solid sodium hydroxide and steam-distilled again. The distillate was extracted with diethyl ether (4 x 15 ml), dried (Na2SO4), and evaporated.
The free amines (6) were treated with equimolar amounts of para-toluenesulphonic acid in ethanol. Crystalline ammonium para-toluenesulphonates (7) were precipitated with diethyl ether and purified by dissolving in a small amount of ethanol and then reprecipitating with an excess of diethyl ether. The yields of the free amines (6) and the mp's and analytical data of the ammonium para-toluenesulphonates (7), are compiled in the Table.
References
[1] E. Fabiano, B. T. Golding, and M. M. Sadeghi, Synthesis, 1987, 190 and references cited therein.
[2] E. Slusarska and A. Zwierzak, Liebigs Ann. Chem., 1986, 402.
[3] A. Koziara, K. Osowska-Pacewicka, S. Zawadzki, and A. Zwierzak, Synthesis, 1985, 202.
[4] 1. M. Downie, J. B. Lee, and M. F. S. Matough, J. Chem. Soc., Chem. Commun., 1968, 1350 and references cited therein.
[5] R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801.
[6] B. A. Clement and R. L. Soulen, J. Org. Chem., 1976, 41, 556; G. Burton, J. S. Eldler, S. C. M. Fell, and A. V. Stachelski, Tetrahedron Lett., 1988, 3003.
[7] A. Hassner and M. Stern, Angew. Chem., Int. Ed. Engl., 1986, 25,478.
(Stranger)
02-26-03 19:06
No 412131
Unless you are going to do a distillation to clean the product up I don't see this as a good method with all the triphenylphospine being produced. A better method would be to make the halide and reduce the azide to the amine with magnesium and methanol (works great and easy to clean up.)
(Hive Bee)
03-06-03 11:37
No 414362
(Rated as: excellent)
This is from a Shulgin article, he prepare these compounds to make some 2C-EF and DOEF with high pressure fluorination.
I also incorporated the preparation of the tosylates, which are useful to swap with azide too. See also ../rhodium /fenflur
Experimental [1]:
Preparation of 2-(2,5-dimethoxyphenyl)ethanol (9):
A solution of sodium borohydride (5.30 g, 139.5 mmol) in THF (50 ml) was cooled (0 °C) and then 2,5-dimethoxyphenylacetyl chloride (15.0 g, 69.76 mmol) in THF
(10 ml) added (dropwise). The resulting pink suspension was stirred at 0 °C (30 min), then warmed to reflux and stirred for 2 h. The solution was cooled (0 °C) and the reaction mixture was quenched with acetone (10 ml) and then water (30 ml ). The solution was filtered and the solvent removed under reduced pressure to yield a crude gold oil.
Kugelrohr distillation of the crude oil afforded the alcohol (9.36g,74%) as a clear oil, b.p.105-107°C/0.5mmHg.
Chimimanie's voice: Another method to 2-(2,5-dimethoxyphenyl)ethanol is from the reaction of the lithio derivative with ethylene oxyde, i have the ref somewhere, a Shulgin article on DOEF too.
Preparation of (+-)-1-(2,5-dimethoxyphenyl)-2-propanol:
1,4-Dimethoxybenzene (15.0 g, 138 mmol) was disolved in THF (40 ml) and cooled to 0°C and n-butyllithium (11.38 ml, 7.65 g, 119.6 mmol, 10.5 M in hexanes) added (slow stream). After addition was complete, the mixtur2e was warmed to reflux and stirred for 48 h. The tan, opaque anion solution was cooled to 0 °C and propylene oxide (8.4 ml, 6.94 g, 58 mmol) in THF (10ml) added (slow stream). The resultant yellow mixture was stirred at 0 °C (1 h), then at 20°C (50 min) followed by heating at reflux for 2.5 h. The reaction mixture was cooled to room temperature and quenched with water (25 ml) to afford a white suspension which was diluted with THF (50 ml) and then water (100ml). The THF was removed under reduced pressure and the resulting aqueous portion was extracted with CH2Cl2 (300ml). The organic portion was washed with brine (50 ml) then dried (MgSO4), filtered and the solvent removed in vacuo to provide a crude gold oil. The oil was purified by column chromatography (ethyl acetate/hexane, 1:5) and the
isolated residue was Kugelrohr-distilled to yield the alcohol (13.32 g, 63%) as a clear viscous oil, b.p. 118-120 °C/0.7 mmHg.
1-(2,5-Dimethoxyphenyl)-2-tosyloxyethane
A mixture of the alcohol (9) (2.26 g, 12.4 mmol) and pyridine (980 mg, 12.4mmol) in CH2C12 (20 ml) was cooled to 0 °C and treated with TsCl (2.37 g, 12.4 mmol). The mixture was stirred at 0°C for 2 h, then stored at 5°C (22h). The mixture was poured into ice water (100 ml) and extracted with CH2Cl2. The organic portion was washed successively with cold (4°C) 1 N HCl, sat. NaHCO3 and then
brine. The organic layer was dried (Na2S04), filtered and the solvent removed under reduced pressure (20°C) to provide a gold oil. Purification of the oil by column chromatography (Et20/hexane,1:4) afforded the tosylate (3.08 g, 74%) as
a clear oil. The tosylates described below were prepared in an
analogous fashion.
1-(2,5-Dimethoxyphenyl)-2-tosyloxypropan
The tosylate was obtained (82%) after HPLC purification (CH2Cl2) as a white solid, m.p. 63-65°C.
Reference:
[1] High pressure nucleophilic fluoride-ion substitution reactions: formation of fluoroalkylbenzenes, John M. Gerdes, Robert N. Keil, Alexander T. Shulgin and Chester A. Mathis, Journal of Fluorine Chemistry 78 (1996) 121-129.
(Chief Bee)
03-15-03 13:13
No 417383
(Rated as: excellent)
Synthesis of 2,5-Dimethoxy-4-Fluoroethylamphetamine (DOEF) (../rhodium /doef.h
(I'm Yust a Typo)
03-15-03 13:31
No 417389
'The facile synthesis of 6 was effected by employing the readily available starting material DOB'
:=)
(Hive Bee)
04-13-03 17:16
No 426454
(Rated as: excellent)
Hi!
Oxidation by mercury(II) salts. I. Hydroxyalkylation of aromatic rings by mercurated ethylene and propylene. M. Julia, E. Colomer and R. Labia Bulletin de la Societe Chimique de France (1972), (11), 4145-8
This is a translation of a french article which describe the synthesis of various dimethoxy-phenylethanols (and propan-2-ols). Those alcohols can be halogenated or tosylated and then swapped with azide/reduction or subjected to the one-pot synthesis described above. The advantage of this method is that it is easy (much easier than lithiation/ethylene oxyde or propylene oxyde) and OTC. The disadvantage is of course the use of mercury salts, which are obviously hazardous. Anyway i like this reaction, it open a new road to 2C-H from dimethoxy benzene in two steps (with the second step beeing the (not OTC) PPh3/CCl4/NaN3 reaction) with an acceptable yield.
This reaction should only bee done by educated individuals, with the skills and knowledge. Mercuric vapors are toxics, mercuric salts are toxics, organomercuric compounds are even more deadly. Metallic mercury is hazardous to the environnement, it should bee recycled to the mercuric acetate for another batch.
They say than direct ammonolyse might bee possible once the organomercuric compound is synthetised, without passing through the alcohol stage.
Fluoboric acid is used here, but it can bee replaced by perchloric acid (but that is more dangerous) or BF3.
Experimental
3,4-dimethoxy-phenyl beta-ethanol acetate
In a 500 mL three necked RBF equipped with a stirrer and a condenser is put 0.1 mol (32g) of mercuric acetate in 150 mL of acetic acid. A stream of ethylene is bubbled in till half an hour after the total dissolution of the mercuric salt. There is then added 0.2 mol (31g) of veratrol and 5 mL of a 40% water solution of fluoboric acid. After 3 hours at 80°C, 20g of metallic mercury is obtained (100%) and the product is extracted with CHCl3. Yield 13.5g (60% based on Hg) of 3,4- dimethoxy-phenylethanol acetate (bp 130-133°C). The acetate is then saponified in ethanolic KOH with a yield of 90%.
The alcoylation of the three dimethoxybenzenes (2,5-, 2,4- and 3,4-) by mercurated ethylene or propylene is done by the same way than with ethylene and veratrol. The six phenylethyl acetates are obtained with yields of 60%.
The six alcohols obtained after saponification are caracterised with their o-naphtyl urethanes or identified with authentic samples.
Dimethoxy phenyl-2 ethanols and their Naphtyl-urethanes
3,4- mp=128°C (urethane); 47°C (alcohol)
2,5- mp=151°C
2,4- mp=134°C; 65°C
Dimethoxy phenyl-propan-2-ols and their Naphtyl-urethanes
3,4- mp=92°C; 40-42°C
2,4- mp=104°C
2,5- no mp given.
when Ar=2,5 dimethoxyphenyl no crystallised naphtyl urethane could be obtained
EDIT:
Another preparation for 2,5-dimethoxyphenyl-propan-2-ol (by lithiation) is in Post 426585 (Antoncho: "Not quite easy, but .....", Russian HyperLab), thank to our Official Russian Translator, Antoncho. Better yield but harder chemistry.
(Hive Bee)
07-29-03 10:38
No 450577
(Rated as: excellent)
This method goes well for recycling mercury into mercury(II) acetate:
Hg metal -> HgSO4:
First, attack Hg metal with conc. H2SO4, like in ../rhodium /mercury
Well, now you have the HgSO4 in H2SO4, let it cool, then pour off the H2SO4, dissolve in water the HgSO4, filter if you find it wise to do so...
HgSO4 -> yellow HgO:
Prepare a solution of NaOH in iced water, and setup an ice bath. Slowly add the cold NaOH solution to the cold mercury sulfate solution, you will see some yellow HgO come then dissolve, after a while, when enough NaOH is added (use a somewhat big excess), you will have the flask full of yellow HgO crystals. When you find the precipitation is finished, filter at buchner, you could dry it with small quantity MeOH then et2O if you want. Dispose properly of the filtrate, and test if there is some remaining mercury in it, by adding a few more NaOH solution.
Yellow HgO -> Mercury(II) acetate: [1],[2]
HgO + 2 CH3COOH = Hg(CH3COO)2 + H2O
MM 216,6 120.1 318.7 18.0
A solution of 20g of yellow HgO in 30 mL of 50% CH3COOH is prepared on a waterbath. It is filtered through a jacketed filter heated with hot water, and the filtrate is cooled with ice. The crystals are suction-dried and washed with ethyl acetate. The product is recrystallized from hot ethyl acetate or from hot water slighty acidified with acetic acid. The salt is dried in a vacuum dessicator over CaCl2.
Some more facts about mercuric acetate: [1],[2]
Use: as a mercurizing and oxidizing agent and for the absorption of ethylene.
Properties: Nacreous, light-sensitive crystalline flakes. On storage acquires a yellow tinge and an odor of CH3COOH (formation of a basic salt). MP: 178-180°C, decompose at higer temperatures.
Solubility: (0°C) 25g, (19°C) 36.4g / 100mL water and about 100g at 100°C with partial dec.). The compound in 0.2N aqueous solution is approximatively 30% hydrolized; the yellow basic salt precipitates on diluting or heating; soluble in ethyl acetate. d 23 3.286.
References:
[1] Wagenknecht, Juza in Handbook of Preparative Inorganic Chemistry, vol. 2, G. Brauer, Ed. (Academic Press, New York, 2nd ed., 1965) pp 1120-1121.
[2] Gmelin-Kraut. Hdb. anorg. Chem., 7th ed., V2, 826, Heidelberg, 1914, modified.
Safety:
-Beware Hg metal vapors.
-Beware Hg salts intake.
-Absolutely beeware organic Hg exposure
Interesting links for toxicity:
http://www.atsdr.cdc.gov/toxprofiles/tp4
Q: And why to not do the direct Hg metal to Hg diacetate transformation with peracetic acid???
A: look DOI:10.1007/s002040050563
The source of methylmercury was most likely inadvertent synthesis. This may have followed the pattern suggested for the Minamata epidemics where the synthesis was also inadvertent. According to this scheme (Irukayama 1977) the first step is the oxidation of acetate to peracetic acid:CH3COOH + O <-> CH3COOOH
and the second step is the formation of methylmercury from peracetic acid and inorganic mercury:CH3COOOH + HgX2 -> CH3HgX + CO2 + HX
Peroxide, inorganic mercury salt and acetate, the essential ingredients of this reaction, were present in the reactor served by the patients. Compared with mercury vapour such a synthesis increased the risk of intoxication because methylmercury acetate is 5.4 times more volatile than elemental mercury (Swensson and Ulfvarson 1863), it is more toxic and the neurological damage is irreversible.
Hell, better to avoid CH3COOOH + HgX2 -> CH3HgX + CO2 + HX then ![]() .
.
(Chief Bee)
06-07-04 17:03
No 512087
(Rated as: excellent)
Magnesium/Hydrazinium Monoformate: A new Hydrogenation System for the Selective Reduction of Nitro Compounds
K. Abiraj, Shankare Gowda, D. Channe Gowda
Synth. React. Inorg. Met.-Org. Chem. 32(8), 1409-1417 (2002) (../rhodium /nitro2
Abstract
The nitro group in aliphatic and aromatic nitro compounds, which also contain reducible substituents such as alkenes, nitriles, carboxylic acids, phenols, halogens, esters, etc., is selectively and rapidly reduced at room temperature to the corresponding amine in good yield by employing hydrazinium monoformate in the presence of magnesium powder. It was observed that, hydrazinium monoformate is more effective than hydrazine or formic acid or ammonium formate and reduction of the nitro group occurs without hydrogenolysis in the presence of low-cost magnesium compared to expensive metals like palladium, platinum, ruthenium, etc.
The Hive - Clandestine Chemists Without Borders
(Hive Bee)
07-06-04 10:44
No 517769
(Rated as: good read)
Reduction of Aromatic Nitro Compounds under Solvent-free Conditions
using Alumina-supported Hydrazine/FeNH4(SO4)2·12H2O
Abstract
Aromatic nitro compounds were easily reduced to the corresponding amino compounds with hydrazine
hydrate supported on alumina in the presence of FeNH4(SO4)2?12H2O
.
Journal of the Chinese Chemical Society, 2004, 51, 569-570
RESULTS AND DISCUSSION
Microwave irradiation has been success fully applied in organic synthesis. Recently, reactions
facilitated by microwaves under solvent-free conditions have attracted more attention be cause
of their enhanced selectivity and milder reaction conditions. Recyclability of the inorganic solid
supportis often possible thus rendering the procedure relatively environmentally acceptable.
In this way, we successfully reduced the aromatic nitro compounds with hydrazine hydrate
supported on alumina in the presence of FeNH4(SO4)2-12H2O.
REFERENCES
1. Han, B. H.; Shin, D. H.; Lee, H. R.; Ro, B. H. Bull. KoreanChem. Soc. 1989, 10, 315.
2. Lee, H. R.; Ryoo, E. S.; Shin, D. H. Taehan Hwhakhoe Chi.1988, 32, 607.
3. Han, B. H.; Shin, D. H. Bull. Korean Chem. Soc. 1985, 6,320.
4. Furst, A.; Berlo, R. C.; Hootom, S. Chem. Rev. 1965, 65, 51.
5. Bavin, P. M. G. Can. J. Chem. 1958, 36, 238.
6. Zhilina, O. D.; Vasilev, A. M.; Shagalov, L. B.; Suvorov, N.N. Khim.Geterotsikl.Geterotsikl.Soedin.1986
7. Yuste, F. Tetrahedron Lett. 1982, 23, 147.
8. Balcom, D.; Furst, A. J. Am. Chem. Soc. 1953, 75, 4334.
9. Hung, Teljes HU 52.755, Chem. Abstr. 1991, 114, 142871f.
10. Hirashima, O.; Manabe, T. Chem. Lett. 1975, 259.
11. Clive, D. L. J.; Angoh, A. G.; Sharon, B. M. J. Org. Chem.
1987, 52, 1339.
12. Hine, J.; Hahn, S.; Miles, D. E.; Ahn, K. J. Org. Chem. 1985,50, 5092.
13. Miyata, T.; Ishino, Y.; Hirashima, T. Synthesis. 1978, 834.
14. Lauwiner, M.; Rys, P.; Wissmann, J. J. Appl. Catal. A. 1998,72, 141.
15. Benz, M.; van der Kraan, A. M.; Prins, R. J. Appl. Catal. A.
1998, 39, 2573.
16. Kumar, P. S.; Sanchez-Valente, J.; Figueras, F. Tetrahedron Lett. 1998, 39, 2573.
17. Han, B. H.; Shin, D. H.; Cho, S. Y. Tetrahedron Lett. 1985,26, 6233.
18. Han, B. H.; Shin, D. H.; Cho, S. Y. Tetrahedron Lett. 1990,31, 1181.
19. Li, Z.-L. Ox i da tion-reduction Re ac tion in Or ganic Sythesis.1985, 104.
20. Eyre; Spottiswoode, I. H.; Bunbury, H. M. Science 1965.
Just hold on to the thread...that keeps us going
http://www.chiapaslink.ukgateway.net/